




Introduction
FRDA is an autosomal recessive degenerative condition with neuronal and non-neuronal manifestations [1]. Cardiomyopathy is detected in two thirds of individuals with FRDA [2-4]. Individuals with FRDA generally present with progressive cardiomyopathy of the left ventricle, which is the leading cause of death in FRDA due to arrhythmias and/or heart failure [5,6]. Little is known of the cellular impacts of FRDA in the heart, but cardiomyocyte necrosis and cellular fibrosis have been identified [6,7]. Systolic function generally remains normal until late in disease progression [8]. In approximately 96% of affected individuals, FRDA is due to homozygosity for an unstable expanded GAA repeat mutation in the first intron of FXN resulting in reduced expression of the nuclear-encoded mitochondrial protein FXN [9-11]. Despite the identification of FXN, its precise role in FRDA pathogenesis remains elusive and remarkably little is known about the molecular pathology of cardiomyocytes in FRDA [12].
Human iPSCs [13-15] have been derived from individuals with FRDA [12,16-19]. Morphological abnormalities and a disorganized mitochondrial network in iPSC-derived- cardiomyocytes have been identified [16,19]. There are no abnormalities under basal conditions, when cultivated in the presence of iron, cellular hypertrophy occurs [19]. However, more detailed functional studies are needed to characterize the cardiomyocytes from FRDA iPSCs. Here, we derived FRDA iPSCs to assess the electrophysiological and calcium (Ca2+) cycling properties of cardiomyocytes, to identify potential mechanisms underlying the cardiomyopathy observed in FRDA.
Results
Generation of three FRDA-iPSC lines and differentiation into cardiomyocytes
We generated FRDA-iPSC lines from three individuals with different GAA length repeat numbers (Table 1). We also collected data regarding disease severity as measured by the Friedreich Ataxia Rating Scale (FARS) [20]. The FARS is scored out of 167, a higher score indicating greater disease severity. Clinical parameters of individuals from whom the lines were derived are as follows: FA6 (female; GAA1 1077, GAA2 1077; FARS score 96.5); FA8 (male; GAA1 476, GAA2 545; FARS score 64.5); FA9 (male; GAA1 733, GAA2 943; FARS score 118). The individuals with FRDA from which iPSCs were derived presented with the following cardiac phenotypes: FA6: normal ejection fraction (55%), normal ventricular wall thickness, mildly dilated left atrium (mild cardiomyopathy); FA8: normal ejection fraction (60%), moderate increase in relative wall thickness (RWT), severe dilatation of the left atrium (typical FRDA cardiomyopathy) and FA9: low normal ejection fraction (50%), increased RWT, borderline increase in left atrial size (typical FRDA cardiomyopathy).
We used nucleofection to deliver episomal vectors containing OCT4, SOX2, KLF4, L-MYC, LIN28, shRNA against p53 and eGFP into fibroblasts. Pluripotent clones were expanded and three clones were selected for each patient (CL1-3). All clones expressed the pluripotency markers OCT4 and TRA-1-60 (Fig.1A-S). The intron 1 FXN GAA expansions were measured for all fibroblasts and iPSC clones (Table 1, Suppl. Fig. 1). As reported for other FRDA iPSCs [16-18], we observed similar repeat numbers as well as contractions and expansions for all lines, with slight variations between clones of the same line (Table 1). Importantly, the patient-derived iPSC lines each maintained the reduced FXN mRNA expression that is characteristic of FRDA, when compared to control cells (Fig. 2A). The iPSCs were karyotypically normal (data not shown), and pluripotent, being able to differentiate into cells of the three germ layers as assessed by embryoid body (EB) formation (Suppl. Fig. 2, Suppl. Fig. 3, Suppl. Fig. 4).
Figure 1. Generation of iPSC lines from FRDA-patients. Immunostaining of FA6 CL1 (A, D), CL2 (B, E), CL3 (C, F) for OCT4 (A-C) and TRA-1-60 (D-F); FA8 CL1 (G, J), CL2 (H, K), CL3 (I, L) for OCT4 (G-I) and TRA-1-60 (J-L); FA9 CL1 (M, P), CL2 (N, Q), CL3 (O, R) for OCT4 (M-O) and TRA-1-60 (P-R). (S) Negative isotype. Cells were counterstained with DAPI (blue). Scale bars: 50 μm.
Table 1. GAA repeats (GAA1/GAA2). GAA1: smaller allele repeats; GAA2: longer allele repeats. F: Female, M: Male. FARS: Friedreich Ataxia Rating Scale.
| FA6 (F, FARS: 96.5) | FA8 (M, FARS: 64.5) | FA9 (M, FARS:118) | |
| Patient, predicted | 1077/1077 | 476/545 | 733/943 |
| Fibroblasts | 854/247 | 481/576 | 788/109 |
| CL1 | 893/281 | 579 | 963 |
| CL2 | 887/273 | 593/323 | 980/383 |
| CL3 | 980/294 | 576/315 | 991 |
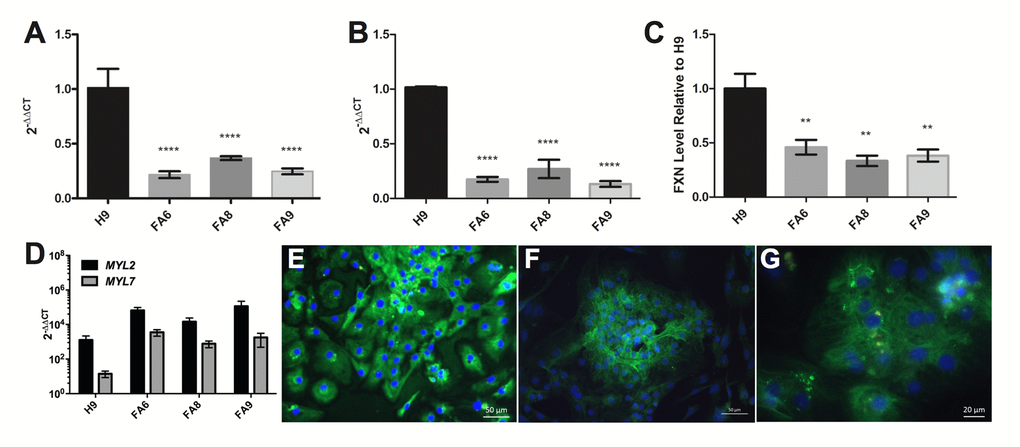
Figure 2. FRDA-iPSCs and - cardiomyocytes retain low levels of FXN and are mainly of ventricular phenotype. (A, B) qPCR and (C) dipstick analysis showing low levels of FXN mRNA (A, B) and protein (C) in undifferentiated cells (A) and their cardiac derivatives (B, C). Significance was assessed by comparing FRDA-iPSCs to undifferentiated H9 controls (A) or FRDA-iPSC derived cardiomyocytes to H9 derived cardiomyocyte controls (B, C). One-way ANOVA followed by Bonferroni’s multiple comparison test, ** p<0.01, **** p< 0.0001. (D) qPCR analysis of cardiomyocytes showing significantly higher expression of MYL2 than MYL7 across all cell lines (p<0.05, paired t-test). (A-D) Data are mean ± SEM of combined clones or 3 individual experiments, normalized to ACTB and relative to undifferentiated cells (A, B, D) or normalized to the control line cardiomyocyte (C). (E-G) Representative images of FA6- cardiomyocytes (E), FA8- cardiomyocytes (F) and FA9- cardiomyocytes (G) for MYL2/MLC2v (green), MYL7/MLC2a (red, weak or absent) and counterstained with DAPI (blue).
All lines were differentiated into cardiomyocytes using a small-molecule based approach [21]. In basal conditions, FRDA- cardiomyocytes were not hypertrophic, as assessed by the absence of nuclear localisation of NFATC4 (Suppl. Fig. 5), a nuclear translocation known to be a marker of hypertrophy [22,23]. The FRDA- cardiomyocytes retained low levels of FXN mRNA and protein (Fig. 2B, C). Phenotypes were assessed at day 35. At this stage, transcripts of MYL2 were significantly higher than MYL7 across all cell lines (Fig. 2D). The cardiomyocytes expressed MYL2/MLC2v protein and MYL7/MLC2a was rarely observed (Fig. 2E-G). Taken together, these data suggest a ventricular phenotype in the cardiomyocytes that were generated with minimal atrial cardiomyocytes for all lines (Fig. 2D-G) [19,24].
FRDA-iPSC-derived cardiomyocytes display phenotypic abnormalities
The Multi Electrode Array (MEA) revealed electrophysiological anomalies in the different FRDA lines, with consistent effects in all three clones of all FRDA lines. All FRDA lines showed similar basal beat rates (30-40 beat per minute, Fig. 3A) which were slower than control cardiomyocytes. All lines showed similar corrected extracellular field potential durations (cFPD, Fig. 3B). We assessed the root of the mean of the sum of the square of the difference in the RR interval (RMSSD), that measures the variation in a consecutive series of intervals between field potentials (Fig. 3C, D). FRDA-iPSC-derived cardiomyocytes showed significantly increased RMSSD under basal conditions and when treated with isoprenaline (10-6 M, Fig. 3C, D). Elevated RMSSD in cultured cardiomyocytes, including those derived from PSCs, is indicative of Ca2+ handling abnormalities. When FRDA- cardiomyocytes were examined under basal conditions or following treatment with isoprenaline, application of nifedipine (10-8 M), which partially blocks L-type Ca2+ channels, prevented an increase in RMSSD, thus confirming that Ca2+ is responsible for the abnormal beat rate variability in line with other studies on PSC-derived cardiomyocytes. In contrast, blocking K+ channels with TEA (10-8 M) did not modify the effect of isoprenaline, with an increase in RMSSD and in cFPD (data not shown). Altogether, these data suggest that FRDA- cardiomyocytes display a significant increase in beat rate variability, demonstrating a potential for cardiac dysfunction, compared to the control cardiomyocytes. These data also suggest that impairment in Ca2+ handling is responsible for the observed electrophysiological phenotype. This was confirmed by assessing Ca2+ transients. In the FRDA-cardiomyocytes significantly lower diastolic and systolic Ca2+ levels and reduced transient amplitude signals were observed compared with control cardiomyocytes (Fig. 3 E-H). Collectively, our data demonstrates a Ca2+ handling impairment in the FRDA cardiomyocytes.
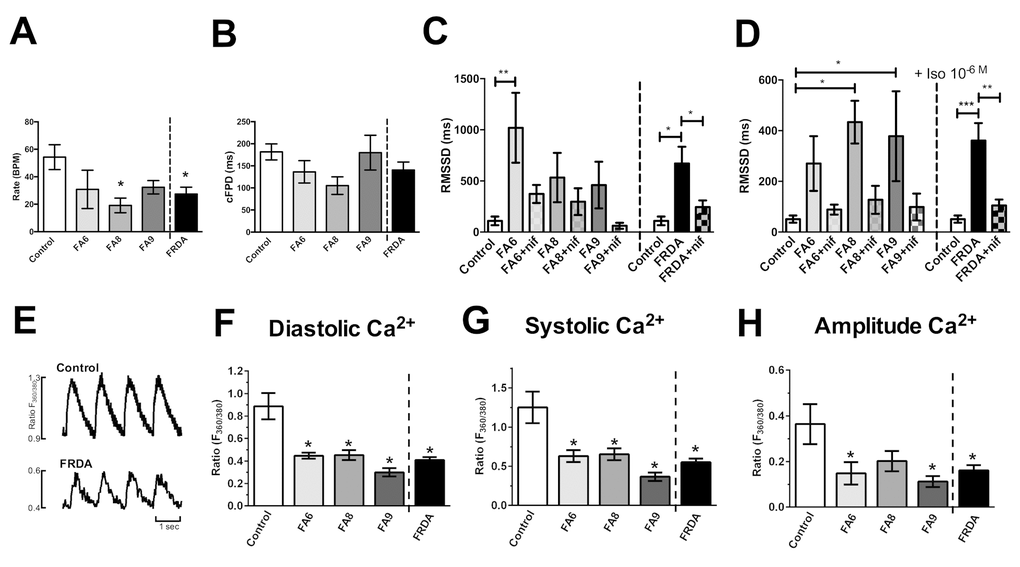
Figure 3. Assessment of phenotypes in FRDA iPSC-derived cardiomyocytes. (A) Beating rates (beats per minute, BPM), (B) corrected extracellular field potential durations (cFPD) and (C, D) the root of the mean of the sum of the square of the difference in the RR interval (RMSSD) (ms). Data shows RMSSD at basal (C) and 10-6M (D) isoprenaline ± nifedipine (NIF). (E) Representative Ca2+ transients. (F) Diastolic, (G) systolic and (H) amplitude Ca2+ in Control and FRDA- cardiomyocytes. (A-D) Data are mean ± SEM of combined values of 3 clones of each line (n=3 per line) and as a pool of all 3 clones of all FRDA lines (n=9 FRDA independent samples) and control cardiomyocytes (n=6). (F-H) Data are mean ± SEM of combined values of control cardiomyocytes (n=5), all 3 clones of each FRDA line (n=8-14 per line) and as a pool of all 3 clones of all FRDA lines (n=35 FRDA independent samples). Statistics: (A-D, F-H) One-way ANOVA followed by Bonferroni’s multiple comparison test, * p<0.05, **, p<0.01, ***, P<0.001.
Discussion
Our data reveal electrophysiological anomalies in FRDA iPSC derived - cardiomyocytes, with an increase beat rate variability. As partial inhibition of L-type Ca2+ channels with nifedipine abolished this electrophysiological phenotype, it demonstrates that the electrophysiological impairment is due to Ca2+ handling abnormalities. As cFPD were not increased in the FRDA iPSC-derived cardiomyocytes, Ca2+ overload is unlikely. This was confirmed with measurement of Ca2+ cycling, which identified that FRDA cardiomyocytes show low Ca2+ transients. Yet, it is difficult to reconcile how the increased beat rate variability is remedied by the Ca2+ channel blocker nifedipine, when the Ca2+ transients suggest that cytosolic Ca2+ levels were already lower than in the controls. It is feasible that in the context of mitochondrial dysfunction in FRDA, ATP-dependent ion channels, particularly sarco/endoplasmic reticulum Ca2+-ATPase, may lack the required ATP for proper functioning, leading to reduced Ca2+ storage in the sarcoplasmic reticulum and therefore reduced Ca2+-induced Ca2+ release during an action potential. There have been two previous reports describing phenotypes in FRDA iPSC-derived cardiomyocytes, including Ca2+ handling deficiencies, albeit in the presence of exogenous iron [19,24]. Yet, whilst iron accumulation is generally regarded as a common feature of FRDA pathogenesis, there is scant evidence of iron accumulation in the heart [25]. The data reported here provide the first demonstration of a phenotypic abnormality in cardiomyocytes derived from FRDA-iPSCs without such artificial iron treatment or external stimulus. This suggests that reduced FXN causes dysfunction in cardiomyocytes prior to events such as accumulation of iron. The establishment of a phenotype, itself reversible by selective treatment with nifedipine, now forms a platform to screen molecules known to modify FXN and directly assess their impact on human FRDA- cardiomyocytes, and hence contribute to the discovery of specific treatments for FRDA. Indeed, cardiomyocytes derived from patient iPSCs show clear phenotypic abnormalities, consisting of an increase in beat rate variability and reduced Ca2+ transients. Although not assessed here, mitochondrial oxidative phosphorylation (OXPHOS) deficiencies, which are reported in FRDA [26], can reduce mitochondrial Ca2+ uptake and ATP production [27,28]. It has also been suggested that mitochondrial OXPHOS defects in FRDA could be an indirect consequence of oxidative stress [29]. Given the impact of reduced Ca2+ levels on mitochondrial function, our data suggests that Ca2+ handling defects could also contribute to the perceived OXPHOS deficiencies. Treatment of FRDA- cardiomyocytes with nifedipine improved phenotypes detected by MEA. Whilst it may seem contradictory that partially inhibiting L-type Ca2+ channels improves cardiomyocytes with low Ca2+ levels, it is possible that nifedipine is acting on other Ca2+ channels independently [30]. This may either mask or restore the Ca2+ levels in the cardiomyocytes [31]. Further in-depth investigations directed at Ca2+ handling machinery of FRDA iPSC-derived cardiomyocytes should unravel the mechanisms behind these phenotypes and identify therapeutic targets.
It might be counterintuitive to relate an increased RMSSD in cultured cardiomyocytes to patient phenotypes. Indeed, patients presenting with cardiomyopathy, including FRDA patients, demonstrate reduced heart rate variability compared to healthy individuals [32]. However, these measures in patients relate more to the function of the autonomic nervous system rather than the function of cardiomyocytes. To our knowledge only two studies have assessed Ca2+ levels in post-mortem FRDA hearts, both describing elevated right ventricle Ca2+ levels, and low-to-normal left ventricle levels in the earlier report versus somewhat elevated left ventricle Ca2+ in a recent report [25,33]. It is however feasible that the FRDA-iPSC derived cardiomyocytes are useful in identifying pathophysiology underlying or preceding the cardiomyopathy observed in FRDA. Importantly, our data clearly indicates that FRDA iPSC- derived cardiomyocytes can be used for screening of compounds able to alter or reverse phenotypes, in human cells, hence providing a novel and unique tool for FRDA research.
Methods
Ethics Committee approvals
All experimental work performed in this study was approved by the Human Research Ethics Committees of the University of Melbourne (0829937, 0605017, 1545383, 1545394) meeting the requirements of the National Health & Medical Research Council of Australia (NHMRC) and conforming to the Declarations of Helsinki.
Biopsies
The biopsy measured approximately 2–3 mm in diameter and was taken via a needle from the inside of the forearm by a qualified clinician. The risks associated with a skin biopsy are small, however the risks of bleeding and infection were minimised through careful technique, the use of antiseptics and sterile instruments. Minimal pain was experienced at the site once the anaesthetic had worn off and none of the individuals reported any complications following this procedure.
iPSC Generation
iPSCs were generated using skin fibroblasts obtained from FRDA (FA6, FA8 and FA9) and control subjects over the age of 18 years by an episomal method as described previously [34]. Reprogramming was performed on passage 8-10 fibroblasts by nucleofection with episomal vectors expressing OCT4, SOX2, KLF4, L-MYC, LIN28 and shRNA against p53 [35].
Maintenance of pluripotent stem cells (PSCs) and cardiomyocyte differentiation
The FRDA-iPSC lines FA6, FA8, FA9 and the control human embryonic stem cell (hESC) line H9 (WiCell) [36] were maintained in the undifferentiated state using TeSR-E8 medium (Stem Cell Tech). H9 was used as a control as the line is very well described; H9-derived cardiomyocytes are structurally and functionally similar to iPSC-derived cardiomyocytes [23,37,38], and have been used as a control for overexpression of mutant MYH7 for modelling cardiomyopathy [23]. hESCs are known to be molecularly and functionally equivalent to iPSCs [39], hence can serve as adequate controls to iPSCs. Embryoid bodies (EB) were obtained as described [40]. In the functional MEA experiments, the control iPSC line, iPSC(Foreskin)-2 [41] was also used, in order to ensure that variations observed between FRDA iPSCs lines and H9 cells were not peculiar to the latter. Differentiation into cardiomyocytes was achieved using a small molecule-based approach, with initial exposure to GSKβ inhibitors (1 µM BIO, 3.5 µM CHIR99021) for 2 days, followed by addition of XAV939 (2 µM) and KY02111 (10 µM) from days 3-8, in IMDM containing 4 mM L-glutamine, 25 mM HEPES, 1% non-essential amino acids, 4 mg/mL human albumin, 100 µM 2-mercaptoethanol, 25 U/mL penicillin, 25 µg/mL streptomycin (all from Thermo Fisher Scientific) and 120 µg/mL L-ascorbic acid sesquimagnesium salt (Sigma Aldrich) [21]. Following two weeks of differentiation in adherent culture, cells were harvested and grown as floating spheres for an additional two weeks in cardiac differentiation medium containing 0.4 mg/mL albumin and 12 µg/mL L-ascorbic acid sesquimagnesium salt. For subsequent work, cells were dissociated with a protease mixture containing 0.1% collagenase I (Wako Pure Chemicals), 0.25% trypsin and 1 U/mL DNase I (Thermo Fisher Scientific) in a buffer consisting of 116 mM NaCl, 20 mM HEPES, 12.5 mM NaH2PO4, 5.6 mM glucose, 5.4 mM KCl, 0.8 mM MgSO4, pH 7.35 (all from Sigma Aldrich). Dissociated cardiomyocytes were plated as monolayers on laminin- or Matrigel-coated plates or slides in albumin- and ascorbic acid-free medium. To assess differentiation, we performed qPCR and/or immunostaining of the cardiac markers actin alpha cardiac muscle 1 (ACTC1), troponin T type 2 (TNNT2), troponin I type 3 (TNNI3) and NK2 transcription factor related locus 5 (NKX2.5) to assess presence of cardiomyocytes, and atrial and ventricular myosin light chain 2 (MLC2a/ MLC2v) to assess the proportion of atrial and ventricular cardiomyocytes. TNN and ACTC staining were also used to assess cardiomyocyte morphology. Nuclear factor of activated T cells 4 (NFATC4) was used to assess the presence of hypertrophy in the cardiomyocytes. A visual assessment of beating cells further confirmed the cardiac phenotype.
Real-time quantitative RT-PCR
Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen, Hilden, Germany), converted to cDNA using High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems, Foster City, CA). Q-PCRs were carried out using TaqMan Universal master mix and the 7900HT Fast Real-Time PCR system using TaqMan gene expression assay for FXN (Hs00175940_m1), MLC2a/MYL7 (Hs01085598_g1), MLC2v/MYL2, (Hs00166405_m1), ACTC1 (Hs01109515_m1), TNNT2 (Hs00165960_m1), TNNI3 (Hs00165957_m1), NKX2.5 (Hs00231763_m1), Glyceraldehyde 3-phosphate dehydrogenase (Human GAPDH, Hs99999905_m1), beta-actin (ACTB; Hs99999903_m1) (all from Applied Biosystems). The relative quantitation was achieved by applying the comparative CT method (ΔΔCT) whereby the mRNA levels were normalized against the level of GAPDH or ACTB and the control group was used as the calibrator.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde (PFA) or ethanol (OCT-4), blocked in 10% fetal calf serum-PBT, and immunostained using the following antibodies: mouse anti-OCT3/4 (Santa Cruz Biotechnology), mouse anti-TRA-1-60 (Millipore), mouse anti-NESTIN (Millipore), rabbit anti-alpha-fetoprotein (AFP, Dako), mouse anti-smooth muscle actin (SMA, R&D systems), mouse anti-ACTC1 (Abcam), rabbit anti-NFATc4 (Santa Cruz), rabbit anti-MYL2 (Proteintech), mouse anti-MYL7 (abcam). Cells were then immunostained with the appropriate conjugated secondary antibodies (Alexa Fluor 568 or 488, Molecular probes-Invitrogen). Nuclei were counter-stained with Hoechst-33342 (Sigma-Aldrich) or DAPI (Invitrogen). Specificity of the staining was verified by the absence of staining in negative controls consisting of the appropriate negative control immunoglobulin fraction (Dako).
GAA expansion analysis
Genomic DNA for GAA expansion analyses was extracted from fibroblasts and iPSCs using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions. The concentration and purity of the genomic DNA were assessed using a Nanodrop1000 spectrophotometer (Thermo Fisher Scientific). The size of the GAA expansion in intron 1 of the FXN gene was determined by PCR using the Expand Long Range dNTPack (Roche, Australia) as recommended with 20 ng template DNA, 0.4 μM of EXP-Bam-F 5′AAGGAAGTGGTAGAGGGTGTTTCACGAGGA3′ and EXP-Bam-R 5′TTTGGATCCAACTCTGCTGACAACCCATGCTGTCCACA3′ primers and 1x Q solution (QIAGEN, Australia). PCR products were electrophoresed on a 1% (w/v) agarose, 1x TAE gel alongside standard DNA markers (200 bp ladder, Promega). Size determination was performed using GeneTools software from SynGene, Synoptics (In Vitro Technologies). The positive control (BAC clone RP11-265B8) and non-expanded alleles in the normal range yielded an 810 bp fragment. The hESC H9, BG01V (ATCC) and the human fibroblast feeders WS1 (ATCC) were included as negative controls for FXN expansion.
Dipstick of FXN expression
Dipstick assay for FXN (Mitosciences) was performed as per the manufacturer’s instructions [42]. This method allows the quantification of FXN by using two specific monoclonal antibodies against different antigens of FXN, in a sandwich ELISA assay. Briefly, one capture antibody bound to a nitrocellulose membrane captures FXN, and a detector antibody conjugated to gold provides a signal when bound to the complex of capture antibody-FXN-detector antibody. Levels of FXN are then detected and quantified by an increased signal intensity.
Electrophysiological characterization
Electrophysiological measurements of cardiomyocytes seeded onto laminin-coated 60 electrodes MEA plates were performed by MEA (Multichannel Systems, Reutlingen, Germany) as previously described [40]. Cells were incubated at 370C in cardiac differentiation medium without albumin and ascorbic acid. Basic receptor and ion channel function were assessed by treatment of cardiomyocytes with nifedipine (inhibitor of L-type Ca2+ channel) and isoprenaline (β-adrenergic receptor agonist) (Sigma-Aldrich). Extracellular field potentials were recorded at baseline and 1 minute after addition of drugs. Data were analyzed offline with MC Rack version 4.3.5 software for beating rate, inter-beat (RR) interval and extracellular field potential duration (FPD). RR interval was defined as the time elapsing between two consecutive beats, and FPD was defined as the time interval between the initial deflection of the field potential and the maximal local T wave. To avoid the influence of beat frequency on FPD, FPD measurements were normalized (corrected FPD, cFPD) with the Bazett’s correction formula: cFPD = FPD/√(RR interval) [43]. Data were expressed as percentage change from baseline with baseline set to 100%. Root of the mean of the sum of the square of the difference in the RR interval (RMSSD) was also assessed.
Intracellular Ca2+ and contractility measurements
Intracellular Ca2+ was measured by microfluorimetry (Ionoptix MA, USA). Cells were loaded with the Ca2+ fluorescent dye FURA-2/AM (5 μM, 10 min incubation at room temperature, Molecular Probes). They were placed in a chamber mounted on the stage of a Motec AE31 inverted fluorescence microscope immersed in medium (IMDM without phenol red, containing 4 mM L-glutamine, 25 mM HEPES, 1% non-essential amino acids, 25 U/mL penicillin and 25 µg/mL streptomycin; the total concentration of Ca2+ in the medium was 1.5 mM), at room temperature and stimulated to contract at 1Hz. Excitation light at 340 nm and 380 nm was provided by a 75-watt xenon lamp and filter wheel. Emitted fluorescence (510 nm) was recorded by a photomultiplier tube, with the output current converted to voltage and digitized for subsequent analysis. Background correction was undertaken at the completion of each cell recording and incorporated into analysis protocol. The following Ca2+ parameters were measured: diastolic Ca2+, systolic Ca2+, amplitude of the Ca2+ transient and time constant of decay of the Ca2+ transient (tau).
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). All statistical analyses and graphical data were generated using Graphpad Prism software (v5.04, www.graphpad.com). Statistical methods utilized were one-way ANOVA followed Tukey’s or Bonferroni’s multiple comparisons test and t-test. Statistical significance was established as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Supplementary Materials
Acknowledgements
The authors would like to thank Matthew J. Bird for his technical help.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Funding
This work was supported by grants from the Friedrich Ataxia Research Alliance (USA, Ireland and Australasia), a National Health and Medical Research Council (NHMRC) Career Development Award Fellowship (AP), a NHMRC-CSL Gustav Nossal postgraduate research scholarship (DEC), a NHMRC Practitioner Fellowship (AWH), Australian Research Council (ARC) Future Fellowships (AP and MD), a Cranbourne Fellowship (RCBW), an ARC special Initiative Stem Cells Australia grant (MD, MPF, AP), a National Stem Cell Foundation of Australia grant (AP) and Operational Infrastructure Support from the Victorian Government.
References
- 1. Delatycki MB, Corben LA. Clinical features of Friedreich ataxia. J Child Neurol. 2012; 27:1133–37. https://doi.org/10.1177/0883073812448230 [PubMed]
- 2. Delatycki MB, Paris DB, Gardner RJ, Nicholson GA, Nassif N, Storey E, MacMillan JC, Collins V, Williamson R, Forrest SM. Clinical and genetic study of Friedreich ataxia in an Australian population. Am J Med Genet. 1999; 87:168–74. https://doi.org/10.1002/(SICI)1096-8628(19991119)87:2<168::AID-AJMG8>3.0.CO;2-2 [PubMed]
- 3. Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996; 335:1169–75. https://doi.org/10.1056/NEJM199610173351601 [PubMed]
- 4. Mottram PM, Delatycki MB, Donelan L, Gelman JS, Corben L, Peverill RE. Early changes in left ventricular long-axis function in Friedreich ataxia: relation with the FXN gene mutation and cardiac structural change. J Am Soc Echocardiogr. 2011; 24:782–89. https://doi.org/10.1016/j.echo.2011.04.004 [PubMed]
- 5. Tsou AY, Paulsen EK, Lagedrost SJ, Perlman SL, Mathews KD, Wilmot GR, Ravina B, Koeppen AH, Lynch DR. Mortality in Friedreich ataxia. J Neurol Sci. 2011; 307:46–49. https://doi.org/10.1016/j.jns.2011.05.023 [PubMed]
- 6. Weidemann F, Störk S, Liu D, Hu K, Herrmann S, Ertl G, Niemann M. Cardiomyopathy of Friedreich ataxia. J Neurochem. 2013 (Suppl 1); 126:88–93. https://doi.org/10.1111/jnc.12217 [PubMed]
- 7. Weidemann F, Rummey C, Bijnens B, Störk S, Jasaityte R, Dhooge J, Baltabaeva A, Sutherland G, Schulz JB, Meier T, and Mitochondrial Protection with Idebenone in Cardiac or Neurological Outcome (MICONOS) study group. The heart in Friedreich ataxia: definition of cardiomyopathy, disease severity, and correlation with neurological symptoms. Circulation. 2012; 125:1626–34. https://doi.org/10.1161/CIRCULATIONAHA.111.059477 [PubMed]
- 8. Kipps A, Alexander M, Colan SD, Gauvreau K, Smoot L, Crawford L, Darras BT, Blume ED. The longitudinal course of cardiomyopathy in Friedreich’s ataxia during childhood. Pediatr Cardiol. 2009; 30:306–10. https://doi.org/10.1007/s00246-008-9305-1 [PubMed]
- 9. Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996; 271:1423–27. https://doi.org/10.1126/science.271.5254.1423 [PubMed]
- 10. Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Dürr A, Mandel JL, et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997; 6:1771–80. https://doi.org/10.1093/hmg/6.11.1771 [PubMed]
- 11. Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet. 1998; 62:111–21. https://doi.org/10.1086/301680 [PubMed]
- 12. Crombie DE, Pera MF, Delatycki MB, Pébay A. Using human pluripotent stem cells to study Friedreich ataxia cardiomyopathy. Int J Cardiol. 2016; 212:37–43. https://doi.org/10.1016/j.ijcard.2016.03.040 [PubMed]
- 13. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007; 318:1917–20. https://doi.org/10.1126/science.1151526 [PubMed]
- 14. Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008; 451:141–46. https://doi.org/10.1038/nature06534 [PubMed]
- 15. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861–72. https://doi.org/10.1016/j.cell.2007.11.019 [PubMed]
- 16. Hick A, Wattenhofer-Donzé M, Chintawar S, Tropel P, Simard JP, Vaucamps N, Gall D, Lambot L, André C, Reutenauer L, Rai M, Teletin M, Messaddeq N, et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis Model Mech. 2013; 6:608–21. https://doi.org/10.1242/dmm.010900 [PubMed]
- 17. Ku S, Soragni E, Campau E, Thomas EA, Altun G, Laurent LC, Loring JF, Napierala M, Gottesfeld JM. Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAA⋅TTC triplet repeat instability. Cell Stem Cell. 2010; 7:631–37. https://doi.org/10.1016/j.stem.2010.09.014 [PubMed]
- 18. Liu J, Verma PJ, Evans-Galea MV, Delatycki MB, Michalska A, Leung J, Crombie D, Sarsero JP, Williamson R, Dottori M, Pébay A. Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev. 2011; 7:703–13. https://doi.org/10.1007/s12015-010-9210-x [PubMed]
- 19. Lee YK, Ho PW, Schick R, Lau YM, Lai WH, Zhou T, Li Y, Ng KM, Ho SL, Esteban MA, Binah O, Tse HF, Siu CW. Modeling of Friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells. Pflugers Arch. 2014; 466:1831–44. https://doi.org/10.1007/s00424-013-1414-x [PubMed]
- 20. Subramony SH, May W, Lynch D, Gomez C, Fischbeck K, Hallett M, Taylor P, Wilson R, Ashizawa T, and Cooperative Ataxia Group. Measuring Friedreich ataxia: interrater reliability of a neurologic rating scale. Neurology. 2005; 64:1261–62. https://doi.org/10.1212/01.WNL.0000156802.15466.79 [PubMed]
- 21. Minami I, Yamada K, Otsuji TG, Yamamoto T, Shen Y, Otsuka S, Kadota S, Morone N, Barve M, Asai Y, Tenkova-Heuser T, Heuser JE, Uesugi M, et al. A small molecule that promotes cardiac differentiation of human pluripotent stem cells under defined, cytokine- and xeno-free conditions. Cell Reports. 2012; 2:1448–60. https://doi.org/10.1016/j.celrep.2012.09.015 [PubMed]
- 22. Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011; 194:355–65. https://doi.org/10.1083/jcb.201101100 [PubMed]
- 23. Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Wang PJ, Nguyen PK, Bers DM, Robbins RC, Wu JC. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013; 127:1677–91. https://doi.org/10.1161/CIRCULATIONAHA.113.001883 [PubMed]
- 24. Lee YK, Lau YM, Ng KM, Lai WH, Ho SL, Tse HF, Siu CW, Ho PW. Efficient attenuation of Friedreich’s ataxia (FRDA) cardiomyopathy by modulation of iron homeostasis-human induced pluripotent stem cell (hiPSC) as a drug screening platform for FRDA. Int J Cardiol. 2016; 203:964–71. https://doi.org/10.1016/j.ijcard.2015.11.101 [PubMed]
- 25. Kruger PC, Yang KX, Parsons PJ, Becker AB, Feustel PJ, Koeppen AH. Abundance and Significance of Iron, Zinc, Copper, and Calcium in the Hearts of Patients With Friedreich Ataxia. Am J Cardiol. 2016; 118:127–31. https://doi.org/10.1016/j.amjcard.2016.04.024 [PubMed]
- 26. Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997; 17:215–17. https://doi.org/10.1038/ng1097-215 [PubMed]
- 27. Visch HJ, Koopman WJ, Leusink A, van Emst-de Vries SE, van den Heuvel LW, Willems PH, Smeitink JA. Decreased agonist-stimulated mitochondrial ATP production caused by a pathological reduction in endoplasmic reticulum calcium content in human complex I deficiency. Biochim Biophys Acta. 2006; 1762:115–23. https://doi.org/10.1016/j.bbadis.2005.09.001 [PubMed]
- 28. Visch HJ, Koopman WJ, Zeegers D, van Emst-de Vries SE, van Kuppeveld FJ, van den Heuvel LW, Smeitink JA, Willems PH. Ca2+-mobilizing agonists increase mitochondrial ATP production to accelerate cytosolic Ca2+ removal: aberrations in human complex I deficiency. Am J Physiol Cell Physiol. 2006; 291:C308–16. https://doi.org/10.1152/ajpcell.00561.2005 [PubMed]
- 29. Orth M, Schapira AH. Mitochondria and degenerative disorders. Am J Med Genet. 2001; 106:27–36. https://doi.org/10.1002/ajmg.1425 [PubMed]
- 30. Curtis TM, Scholfield CN. Nifedipine blocks Ca2+ store refilling through a pathway not involving L-type Ca2+ channels in rabbit arteriolar smooth muscle. J Physiol. 2001; 532:609–23. https://doi.org/10.1111/j.1469-7793.2001.0609e.x [PubMed]
- 31. Visch HJ, Rutter GA, Koopman WJ, Koenderink JB, Verkaart S, de Groot T, Varadi A, Mitchell KJ, van den Heuvel LP, Smeitink JA, Willems PH. Inhibition of mitochondrial Na+-Ca2+ exchange restores agonist-induced ATP production and Ca2+ handling in human complex I deficiency. J Biol Chem. 2004; 279:40328–36. https://doi.org/10.1074/jbc.M408068200 [PubMed]
- 32. Pousset F, Kalotka H, Durr A, Isnard R, Lechat P, Le Heuzey JY, Thomas D, Komajda M. Parasympathetic activity in Friedrich’s ataxia. Am J Cardiol. 1996; 78:847–50. https://doi.org/10.1016/S0002-9149(97)89245-4 [PubMed]
- 33. Huxtable RJ, Johnson P, Lippincott SE, Barbeau A. Free amino acids and calcium, magnesium and zinc levels in Friedreich’s ataxia. Can J Neurol Sci. 1984; 11:616–19. https://doi.org/10.1017/S0317167100035150 [PubMed]
- 34. Hung SS, Van Bergen NJ, Jackson S, Liang H, Mackey DA, Hernández D, Lim SY, Hewitt AW, Trounce I, Pébay A, Wong RC. Study of mitochondrial respiratory defects on reprogramming to human induced pluripotent stem cells. Aging (Albany NY). 2016; 8:945–57. https://doi.org/10.18632/aging.100950 [PubMed]
- 35. Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, Shibata T, Kunisada T, Takahashi M, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011; 8:409–12. https://doi.org/10.1038/nmeth.1591 [PubMed]
- 36. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998; 282:1145–47. https://doi.org/10.1126/science.282.5391.1145 [PubMed]
- 37. Mandel Y, Weissman A, Schick R, Barad L, Novak A, Meiry G, Goldberg S, Lorber A, Rosen MR, Itskovitz-Eldor J, Binah O. Human embryonic and induced pluripotent stem cell-derived cardiomyocytes exhibit beat rate variability and power-law behavior. Circulation. 2012; 125:883–93. https://doi.org/10.1161/CIRCULATIONAHA.111.045146 [PubMed]
- 38. Gherghiceanu M, Barad L, Novak A, Reiter I, Itskovitz-Eldor J, Binah O, Popescu LM. Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: comparative ultrastructure. J Cell Mol Med. 2011; 15:2539–51. https://doi.org/10.1111/j.1582-4934.2011.01417.x [PubMed]
- 39. Choi J, Lee S, Mallard W, Clement K, Tagliazucchi GM, Lim H, Choi IY, Ferrari F, Tsankov AM, Pop R, Lee G, Rinn JL, Meissner A, et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol. 2015; 33:1173–81. https://doi.org/10.1038/nbt.3388 [PubMed]
- 40. Lim SY, Sivakumaran P, Crombie DE, Dusting GJ, Pébay A, Dilley RJ. Trichostatin A enhances differentiation of human induced pluripotent stem cells to cardiogenic cells for cardiac tissue engineering. Stem Cells Transl Med. 2013; 2:715–25. https://doi.org/10.5966/sctm.2012-0161 [PubMed]
- 41. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007; 318:1917–20. https://doi.org/10.1126/science.1151526 [PubMed]
- 42. Bird MJ, Needham K, Frazier AE, van Rooijen J, Leung J, Hough S, Denham M, Thornton ME, Parish CL, Nayagam BA, Pera M, Thorburn DR, Thompson LH, Dottori M. Functional characterization of Friedreich ataxia iPS-derived neuronal progenitors and their integration in the adult brain. PLoS One. 2014; 9:e101718. https://doi.org/10.1371/journal.pone.0101718 [PubMed]
- 43. Zwi L, Caspi O, Arbel G, Huber I, Gepstein A, Park IH, Gepstein L. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009; 120:1513–23. https://doi.org/10.1161/CIRCULATIONAHA.109.868885 [PubMed]