Introduction
The mechanisms involved in ageing and longevity are
currently receiving a great deal of attention, perhaps reflecting the impending
socioeconomic impacts of a more ‘elderly' society. Recent research has revealed
roles for the protein kinase termed ‘the target of rapamycin' (TOR) in
modulating lifespan, and for two of the processes which TOR regulates, i.e.
protein synthesis and autophagy. Studies in diverse model organisms have shown
that impairment of TOR signaling leads to increased life span.
The mammalian target of rapamycin, mTOR,
like its orthologs in other eukaryotes, is a multidomain protein kinase that
interacts with other proteins to form two main types of complex, mTOR complexes
1 and 2 (mTORC1 and mTORC2) (inset Figure 1). Signaling through mTORC1 is much
better understood than signaling through mTORC2: mTORC1 is an important node in
cellular regulation impacting on cell growth that is linked to ageing [1]. Signaling
through mTORC1 is activated by hormones,
mitogens and growth factors, requires
amino acids, and is negatively regulated by stressful conditions, such
as decreased energy (ATP) availability. While both mTORC1 and mTORC2 contain
mLST8 (also termed GβL), mTORC1 associates with raptor, which binds to proteins
that are direct substrates for mTORC1 and mTORC2 binds to rictor. Although
TOR's name reflects the fact that rapamycin can inhibit TOR function via an association of FKBP12 with rapamycin which binds to the
FKBP12-Rapamycin Binding (FRB) domain of TOR,
only (m)TORC1 is sensitive to this drug in the short-term, and not all
functions of this complex are blocked by rapamycin. Dysregulation of mTORC1
signaling contributes to several human diseases - e.g., cancers, cardiac
hypertrophy and tuberous sclerosis [2-4]. Indeed
rapamycin, or its analogs (rapalogs), are in clinical use or trials for a
number of diseases.
mTORC1 regulates a range of essential cellular
functions, the best understood of these being protein synthesis (mRNA
translation), which is positively regulated by mTORC1 (Figure 1) [5]. Conversely,
mTORC1 signaling impairs autophagy [6], a
degradative process through which proteins and other macromolecules are broken
down. This article focuses on the fact that both protein synthesis and
autophagy are implicated in regulating lifespan and ageing and investigates how
mTORC1 links into the ageing signaling network.
1) Protein synthesis
Two widely-conserved effectors of mTORC1 which are
linked to the translational machinery and its control are eukaryotic initiation
factor (eIF) 4E and the kinases that phosphorylate S6, a component of the small
(40S) ribosomal subunit (termed S6Ks). The physiological function of the
phosphorylation of S6 [7] and the
other S6K substrates functionally associated with mRNA translation requires
clarification. However, the function of eIF4E is far better understood. eIF4E
binds the cap structure found at the 5'-end of cytoplasmic mRNAs and also
interacts with other proteins, in particular the multidomain scaffold protein eIF4G [8]. eIF4G, in
turn, interacts with several other proteins. One of these is the
poly(A)-binding protein (PABP). The interaction of eIF4G with eIF4E and PABP
circularizes the mRNA (by bringing together its 5'- and 3'-ends), which
markedly enhances its translation. eIF4G also, indirect-ly, recruits 40S
subunits to the mRNA to initiate translation at the 5'-end. eIF4E and its
interaction with eIF4G are therefore considered to be very important for the
initiation of mRNA translation. The eIF4E:eIF4G interaction is blocked by the
interaction of eIF4E with small phosphoproteins termed eIF4E-binding proteins,
4E-BPs. The best understood of these is mammalian 4E-BP1, which is directly
phosphorylated by mTORC1: this leads to its release from eIF4E, allowing eIF4G
and its partners to bind [8]. In this
way, mTORC1 signaling positively regulates eIF4E activity.
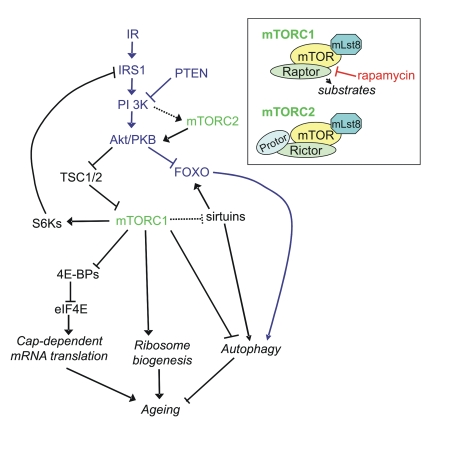
Figure 1. Signaling pathways linking mTORC1 and mTORC2 to ageing via protein synthesis and autophagy. It should be noted that many links have been made
in C. elegans and D. melanogaster and the reader is referred
to Table 1 for homologs of the mammalian proteins in this figure. Inset:
components of mTORC1 and mTORC2.
Table 1. List of autophagy and protein synthesis homologs referred to in this review in S. cerivisiae, mammals, C. elegans and D. melanogaster and their effects on lifespan when known.
| Mammalian | S.
cerivisiae | C. elegans | D. melanogaster | Function | Effect on
longevity |
|
Unc-51 like
kinases 1 & 2 (ULK1&2)
|
Atg1
|
Unc-51
|
DrATG1
|
Induction of
autophagy
|
Loss of function
mutation increases tissue ageing & decreases lifespan in C. elegans
[49]
|
|
APG5
|
Atg5
|
Atgr-5
|
DrATG5
|
Autophagosome
assembly
| |
|
Beclin-1
|
Atg6
|
Bec-1
|
DrATG6
|
Autophagosome
assembly
|
Loss of function
mutation increases tissue ageing & decreases longevity in C. elegans
[49]
|
|
APG7
|
Atg7
|
Atgr-7
|
DrATG7
|
Autophagosome
assembly
|
Knockdown/mutation
reduces lifespan in D. melanogaster and C. elegans [49, 50]
|
|
LC3 (APG8)
|
Atg8
|
Igg-1
|
DrATG8
|
Autophagosome
assembly
|
Reduced
expression decreases longevity & enhanced expression increases lifespan
in D. melanogaster [54,55]
|
|
APG12
|
Atg12
|
Igg-3
|
DrATG12
|
Autophagosome
assembly
| |
|
WIPI1
|
Atg18
|
Atgr-18
|
CG11975
|
Recruitment of
protein to vesicle membrane
|
Loss of function
mutation increases tissue ageing and decreases lifespan in C. elegans [49]
|
|
mTOR
|
TOR
|
Let-363
|
DrTOR
|
Repression of
autophagy
|
Inhibition
extends lifespan in yeast, C. elegans and D. melanogaster [20-22]
|
|
S6K
|
Sch9p
|
Rsks-1
|
dS6K
|
Phosphorylates
S6, a component of the 40S ribosomal subunit
|
Impairing
expression in C. elegans & D. melanogaster extends lifespan
[37, 39]
|
|
eIF4E
|
CDC33
|
Ife-1 - Ife-5
|
eIF4E-4
|
Translation initiation
(binds mRNA's 5'-cap)
|
Knockout in C.
elegans extends lifespan independent of Daf-2 & TOR pathways [39]
|
|
Mnk1
and Mnk2
|
-
|
Mnk-1
|
Lk6
|
eIF4E kinase
|
Affects lifespan in D. melanogaster [43]
|
|
FoxA 1, 2 &
3
|
PHA-4
|
Pha-4
|
forkhead
|
Transcription
factor
|
Enhanced
activity increases lifespan [71]
|
|
FOXO 1, 3 and 4
|
-
|
Daf-16
|
dFOXO
|
Transcription
factor
|
Overexpresssion
increases lifespan in D. melanogaster and C. elegans |
Proper accuracy and control of protein synthesis is
essential. For example, the accumulation of mistranslated and potentially
misfolded proteins can lead to neurodegeneration [9].
Furthermore, protein synthesis is also a costly process, consuming both amino
acids and energy. Indeed, the proportion of cellular energy used in protein
synthesis is estimated to be as high as 30-40% of total ATP (and GTP) [10,11]. This
consideration is important not only with respect to the overall cellular energy
‘economy' but also because the production of ATP in mitochondria is associated
with the generation of reactive oxygen species (ROS) which may have damaging
effects on cellular components. Interestingly, mTORC1 signaling plays a role in
regulating mitochondrial function [12-14]. A
number of studies in a broad range of organisms have now demonstrated links
between mTOR, protein synthesis and ageing/life span. The overall thrust of
these studies is that inhibiting protein synthesis or (m)TOR signaling can
extend life span.
2) Autophagy
Autophagy is a second key process that is
regulated by mTORC1 (Figure 1). Autophagy is a process by which cargo, such as
long-lived proteins and cytoplasmic organelles, is sequestered and delivered to
the lysosomes. Based on the cellular mechanisms of cargo delivery to lysosomes,
three different types of autophagy have been described in mammalian cells [15].
Chaperone-mediated autophagy (CMA) is a form of autophagy wherein a soluble
pool of cytosolic proteins is targeted to lysosomes for selective degradation [16]. Cytosolic
proteins with a CMA-targeting motif bind to a receptor protein, the
lysosomal-associated membrane protein (LAMP-2A), are translocated across the
membrane and are degraded within the hydrolase-rich lumen. In addition to CMA,
there are two other types of autophagy, macro-and microautophagy, which involve
mainly non-selective engulfment of cytosolic regions, including organelles and
soluble proteins. Macroautophagy is the most extensively characterized process
where de novo-formed limiting membranes sequester regions of the cytosol ("bulk
degradation"), but also selectively sequestrate cellular organelles and protein
aggregates into autophagosomes. Such double-membrane vesicles (autophagosomes)
then acquire proteases that are responsible for degrading engulfed material via
a fusion event with endosomes and lysosomes. The formation and fusion of the
autophagic compartment with lysosomes is regulated by a protein-to-protein and
a protein-to-lipid conjugation controlled by the beclin-VPS34 (vacuolar protein
sorting) and the mTORC1 intracellular kinase complex.
Although the process of autophagy is still poorly
characterized (at least in mammalian cells), much progress has been made since
yeast genetic studies identified the first autophagy-related genes (ATGs) about
10 years ago. Now, more than 30 such genes have been discovered. Most of these
are conserved throughout eukaryotes. It is well established that normal
cellular function relies on surveillance mechanisms, molecular chaperones and
proteolytic systems and that many of these functions decline with age. It is
evident that the build-up of damaged cellular components in ageing cells and
tissues is, at least partly, attributable to a decline in autophagy, including
CMA [17,18]. This
has been proposed to be due to a decrease in the clearance of vacuoles, for
example the accumulation of lipofuscin with age in the lysosome could diminish
lysosomal function or the lysosome could be damaged by toxic protein products [19]. A decrease
in the sensitivity of autophagy to regulation by insulin and glucagon has also
been suggested to play a role in modulation of autophagy with age [20,21].
There is extensive evidence that macroautophagy
(referred to as autophagy from here on) is negatively regulated by mTORC1 (eg [22] and
references therein). However, the mechanisms by which mTORC1 controls autophagy
are not well established. The mechanisms regulating the initial stages of
autophagy, at least, appear to involve mTOR-dependent phosphorylation of ULK1
(UNK-51-like kinase), a mammalian serine/threonine protein kinase (Atg1 in
yeast) which forms a 3-MDa complex with Atg13 and FIP200 [23-26]. It has
also been suggested that mTOR acts on ATG genes through the regulation of the
phosphatase PP2A [27]. Less
direct ways by which mTOR may regulate autophagy are via S6K and its
transcriptional targets or signaling through Akt (Figure 1). For example,
using RNAi knockdown in cell lines, S6K has recently been shown to be required
for the starvation-induced autophagic response [28].
In this review we concentrate on TOR dependent
regulation of autophagy, but it should be noted that autophagy is also
regulated in a TOR independent manner, either via IP3 [29] or via
other protein kinases. For example, cAMP-dependent protein kinase (PKA) was
shown to inhibit the induction of autophagy in yeast [30] and Atg1,
13 and 18, which are required for autophagy, are PKA substrates [31].
Furthermore, the AMP-activated protein kinase (AMPK), an important energy
sensor, regulates the phosphorylation of the cell-cycle regulator, p27. p27
can maintain autophagy in a human cell line [32] and its
regulation is proposed to be an important determinant of whether a cell enters
a survival pathway (via autophagy) or apoptosis. All these effectors of
autophagy, including TOR and ATG genes, have been linked with longevity. For a
recent in-depth review on the molecular mechanisms by which autophagy genes
interact with longevity pathways in diverse organisms the reader is also
referred to the paper by Vellai [33].
3) TOR-dependent regulation of ageing via protein
synthesis and autophagy
Recently, it has been proposed that the main driver of
ageing is TOR signaling rather than ROS [34]. Inhibition
of the TOR pathway extends lifespan in yeast, worms and flies [35-37] and a
recent, notable study showed that rapamycin, an inhibitor of mTOR, fed late in
life extends lifespan in genetically heterogeneous mice [38]. Below, we
discuss the ways in which protein synthesis and autophagy may contribute to the
regulation of lifespan by TOR. We summarize drug studies in cells and animals
as well as evidence obtained by genetic analyses in various model organisms.
For information on gene homologues and
their effects on longevity across model organisms the reader is referred to
Table 1.
3.1) mTOR, translation, and life-span
In the nematode worm C. elegans,
several studies have shown that decreasing the amount of proteins involved in
mRNA translation (ribosomal proteins, initiation factors) extends life span -
examples of the latter include eIF4E and eIF4G as well as eIF2 and eIF2B. Both
eIF4E and eIF4G can be controlled by mTORC1 [39,40].
Reduced expression of TOR or S6 kinase also led to longer life. It has been
argued that decreased protein synthesis rates might extend lifespan by reducing
energy consumption and thus diminishing respiration and ROS production.
However, reducing translation still increased lifespan in animals with
decreased respiration, suggesting that protein synthesis affects life span
independently of effects on energy usage or oxygen consumption. Other data
indicate that the effects of decreased mTORC1 signaling may be mediated through
reduced mitochondrial oxidative metabolism (e.g. [41] and
discussion therein). There appears to be a complex interplay between
mitochondria and (m)TOR, with signaling proceeding in both directions between
them [13]. mTORC1
signaling promotes the transcription of genes involved in mitochondrial
function [12] likely as
part of a programme of events to generate the energy required by anabolic
processes such as protein synthesis. Interestingly, the effect of yeast TORC1
on mitochondrial function to influence chronological life span (CLS, the
period in which yeast cells remain viable in a non-dividing state) occurs not
via mitochondrial bio-genesis, but primarily through translational regulation
of OXPHOS complexes [14].
The role of eIF4E in longevity has been the subject of
several recent studies. C. elegans possesses several isoforms of eIF4E,
with different patterns of expression and differing specificities for the
different 5'-cap structures found in this organism. The product of the ife-2
gene is widely expressed in somatic tissues and binds to both the 7-monomethyl
and 2,2,7-trimethyl guanosine cap structures found in mRNAs in this species.
Knockout of this gene has been shown to extend lifespan, in a manner distinct
from the DAF-16 (FOXO) pathway (Figure 1) [39,42], and
enhances resistance to oxidative stress, UV irradiation and heat shock, as well
as to starvation. It should be noted that in the study of Hansen and colleagues
[39] an ife-2
mutant did not extend lifespan in a daf-16 mutant background, implying that the
extension of lifespan due to loss of ife-2 may be DAF-16 dependent.
Interestingly, both increasing and decreasing the levels of the eIF4E kinase,
Lk6, increases lifespan in Drosophila [43].
It remains to be established whether manipulations
that inhibit general protein synthesis affect longevity due to the decreased
rates of overall protein synthesis or perhaps to the concomitant upregulation
of the translation of specific mRNAs whose products help confer resistance to
stress. A combination of these effects may, of course, be involved. The effects
of manipulating eIF4E activity may be especially relevant here, since eIF4E is
involved in cap-dependent translation while many mRNAs for ‘stress' proteins
are encoded by mRNAs by mechanisms that either show a low requirement for the
cap-dependent translation machinery or are translated by cap-independent
mechanisms such as internal ribosome entry segments (IRESs) [44]. Since
longevity is promoted by decreasing the levels of initiation factors required
for general translation (eIF2β[39] or eIF2Bδ[40]) or of
ribosomal proteins [39], it seems
that impairing general protein synthesis, rather than specifically inhibiting
cap-dependent translation, extends lifespan.
The regulation of eIF4E's activity by 4E-BPs (TOR
substrates) could provide a mechanism by which TOR controls longevity. However,
no structural ortholog of the 4E-BPs has been identified in C. elegans
although it is possible that functional orthologs do exist. However, knocking
down TOR extends lifespan further in ife-2 deleted worms, indicating
that the effects of TOR and ife-2 are mediated through distinct
mechanisms [42], further
discussed in section 4.2. Thus, TOR's effects on lifespan in the worm model
appear to operate by additional mechanisms. For example, TOR signaling promotes
ribosome biogenesis, a costly process that is needed for cell growth and
division, across the eukaryota from yeast to mammals [45]. Since the
number of ribosomes in a cell governs the cellular capacity for protein
synthesis, decreased ribosome production may be involved in extending lifespan
in animals where TOR signaling is downregulated.
3.2) Autophagy and the modulation of life-span
As mentioned above, inactivation of TORC1 via treating
cells with rapamycin or nitrogen starvation induces autophagy [46,47]. The
extension of lifespan due to TOR inhibition could potentially occur solely via
TOR's effects on protein synthesis (see above). However, recent studies in C.
elegans suggest a direct role for autophagy in modulating longevity, as
inactivation of autophagy genes (bec-1, unc-51, atg-18)
specifically prevents inhibition of TOR activity from extending lifespan [48,49]. This
indicates that TOR and autophagy act via the same signaling pathways to affect
lifespan.
A recent study using loss of function mutational
analysis in C. elegans showed a clear acceleration in tissue ageing and
a reduced lifespan in worms with loss of function mutations in the autophagy
genes, bec-1, unc-51 and atg-18 [49]. This shows
the importance of autophagy in regulating normal lifespan. These findings
support an earlier study using RNAi knockdown of atg-7 which reduced
lifespan of wild-type C. elegans [50]. Some other
experiments in C. elegans using RNAi knockdown of atg-7 or bec-1,
however, showed no significant effects on lifespan [48,50,51].
These negative findings could be explained by the finding that residual Atg
activity can still produce significant autophagic flux (e.g. in a mammalian
cell system) [52,53]. In Drosophila,
mutation of Atg7 and reduced expression of Atg8 each decreased
longevity [49,54] and
upregulation of the autophagy gene Atg8 increased longevity [55]. Also in
yeast, autophagy has been implicated in ageing (reviewed in [33]). For
example, a recent study showed that the CLS in yeast is reduced by deletion of Atg1
or Atg7, both required for autophagy in yeast [56]. Therefore, loss of autophagy controlled by TOR
accelerates ageing. This is not unexpected because of the importance of
autophagy in maintaining a healthy cellular environment where damaged proteins
and organelles can be eliminated.
4) The role of mTOR in ageing controlled by dietary
restriction and insulin signaling
4.1) Insulin signaling
The effect of TOR on lifespan operates
downstream of the insulin signaling pathway (Figure 1), as indicated by the
observation that the increase in lifespan in an insulin signaling mutant cannot
be further extended by mutations in components of the TOR pathway [21]. A link
between insulin signaling, ageing and autophagy was initially described when
studies in rodent liver showed that an increase in insulin with age causes an
inhibition of autophagy and that the ability of glucagon to upregulate
autophagy is reduced with increasing age [57,58]. More
recently, FOXO (Daf-16) has been shown to directly control the transcription of
autophagy genes, including members of the Atg8 family (LC3, Gabarapl1)
and regulators of autophagy, Bnip3, and Atg12l [59,60].
Upregulation of FOXO also induces autophagy in Drosophila [61],
C. elegans [59] and mouse
muscle fibres [60]. In
addition, a mutation of FOXO caused a reduction of starvation-induced autophagy
in the fat body of Drosophila [61]. The C.
elegans study showed that the upregulation of autophagy in skeletal muscle
via Daf-16 was independent of mTOR, as demonstrated by inhibition of mTOR by
rapamycin or knockdown [59]. Knockdown
of a component of mTORC2 (rictor) did, however, result in FOXO-mediated
induction of autophagy. The authors explain the apparent discrepancy between the
lack of effect of mTOR inhibition and the positive effect of mTORC2 inhibition
on autophagy by a negative feedback of S6K on Akt/PKB activity. It has indeed
become apparent that Akt signaling can be both positively and negatively
regulated by mTOR, depending on the TOR complex. As described above, S6K is a
target of mTORC1. S6K phosphorylates IRS1 at inhibitory sites, inhibiting
activation of Akt [62,63],
upregulating autophagy. On the other hand, mTORC2 has been shown to
phosphorylate S473 of Akt, hence activating Akt and downregulating autophagy [64]. This is
consistent with observations in the Drosophila fat body, whereby
signaling through TOR and PI3K is necessary and sufficient to suppress
starvation-induced autophagy and yet S6K promotes autophagy [65]. The
balance between mTORC1 and mTORC2 signaling therefore could be critical in the
regulation of Akt and hence autophagy and ageing.
Further evidence linking insulin signaling with
autophagy comes from a mouse with targeted deletion of PTEN, in the liver. PTEN
is a lipid phosphatase that reduces PI3K activity and hence an antagonist of
insulin signaling, whose elimination will result in activated Akt and thus
mTORC1. Autophagic degradation in the liver of this mouse was significantly
reduced [66]. In C.
elegans, downregulation of the autophagy gene Bec1 inhibited the longevity
phenotype of the Daf-2 insulin receptor mutant [67], indicating
that the extension of lifespan due to alterations in insulin signaling may
occur, at least in part, via autophagy.
4.2) Dietary restriction and sirtuins
Another well-established mechanism for promoting
longevity is dietary restriction. Dietary restriction in many organisms,
including Drosophila, C. elegans and rodents, is known to induce
autophagy [65,68,69],
which is to be expected given that TOR signaling is impaired when amino acids
levels are low. On the other hand, it has been demonstrated that the decline
in autophagy with increasing age can be prevented by caloric restriction in
mice [70]. Two
autophagy genes, bec-1 and atg-7, have been shown to be required
for the longevity phenotype of the inherent dietary restriction C. elegans mutant
eat-2 [48,51]
indicating that autophagy is required for the lifespan extension induced by
dietary restriction.
The FoxA family of transcription factors is involved
in multiple physiological processes including the regulation of longevity in
response to dietary limitation and related manipulations [71]. Sheaffer
and colleagues identified the AAA+ ATPase ruvb-1 as a component of the
TOR signaling pathway and as a negative regulator of the FoxA homolog pha-4
in C. elegans. They showed that the effects on lifespan of inactivating
TOR or the S6K homolog rsks-1 requires pha-4, whereas the lifespan-promoting
effect of mutations in eIF4E/ife-2 does not require pha-4. This
suggests that eIF4E and TOR affect longevity via distinct mechanisms.
Therefore, nutrient availability may control longevity by affecting TOR
signaling and repressing or enhancing pha-4/FoxA function.
The activation of autophagy due to dietary restriction
in C. elegans was also shown to require PHA-4 (FoxA) activity [48]. Pha-4
is required for the induction of increased numbers of autophagic vesicles under
certain conditions [72], suggesting
that changes in gene expression are required for this process (since pha-4
is a transcription factor). In addition to repressing pha-4, TOR and ruvb-1
regulate the nucleolar accumulation of components, termed box C/D snoRNPs that
are involved in the maturation of rRNAs (which are made in the nucleolus).
Impairing TOR/ruvb-1 signaling is thus expected to interfere with
ribosome production, likely explaining the decreased rates of protein synthesis
seen in worms where TOR signaling or box C/D snoRNP function is perturbed [71]. It remains
to be elucidated which functions of pha-4 are involved in regulating
lifespan.
Data from yeast point to a role for
Sch9p, the probable yeast ortholog of S6K (and which is downstream of TORC1) in
lifespan extension due to dietary restriction [73]. Impairing
S6K/Sch9p activity (using an inactive ‘dominant negative' mutant) also extends
lifespan in Drosophila [37].
Interfering with S6K expression in C. elegans also extended lifespan [39]. S6K has
been implicated in ribosome biogenesis, in oxidative phosphorylation [14], in the
regulation of nucleolar rDNA transcription [45] and in the
control of the translation of mRNAs for ribosome proteins (although recent work
has revealed that S6Ks are dispensable for the latter (reviewed in [7]). It
therefore remains to be established how S6K orthologs affect lifespan.
Studies in yeast implicate TOR-mediated regulation of
proteins called sirtuins in the control of replicative lifespan. Sirtuins are
NAD+-dependent deacetylases that stabilize the rDNA locus and,
interestingly, are also involved in the control of lifespan by caloric
restriction, not only in yeast, but also in C. elegans and in
Drosophila [74,75]. TOR
may impair sirtuin activity through the induction of the Pnc1p nicotinamidase
via the transcription factors Msn2p and Msn4p, whose nuclear localization is
inhibited by TORC1 signaling [74]. Recently a
role for Sirt1 (Sir2 in yeast) in upregulating autophagy was revealed [76]. In this
study Sirt1 was upregulated in mice subjected to starvation and was necessary
for the induction of starvation-induced autophagy. Sirt1-/- mouse
fibroblasts were unable to stimulate basal rates of autophagy and Sirt1
interacted with Atg5, 7 and 8. In yeast, TOR inhibition has been shown to
extend lifespan by increasing Sir2 activity, the same mechanism thought to be
involved in extending lifespan in response to caloric restriction [74]. Recent
work in Drosophila shows that dSir2 interacts with and deacetylates p53,
which mediates, at least in part, the lifespan extending effects of dietary
restriction [77].
Therefore, sirtuins provide an important link between dietary restriction, TOR
signaling and ageing, a link which may arise due to the regulation of autophagy
by sirtuins, via deacetylation and hence activation of FOXO [75] (see
signaling diagram) and/or by direct deacetylation of autophagy components [76]. Given that
sirtuins control processes that defend cells against oxidative stress which, in
turn, can be regulated by TOR (see below), it is noteworthy that SIRT3
physically interacts with the daf-16 homolog FOXO3 within mitochondria [78], organelles
that are signal integrators of oxidative metabolism and perhaps ageing.
5) The interplay of TOR, oxidative/mitochondrial
metabolism and ageing
The relationships between TOR signaling and oxidative
and/or mitochondrial metabolism are complex. While ROS or peroxides induced by
growth factors or UV activate mTOR (e.g. [79-83]),
exogenous hydrogen peroxide inhibits mTOR (>100 μM) [84]. mTOR
itself has been shown to both increase [85] and
decrease [86] ROS levels.
However, these studies were performed on transformed cell lines or
LPS-stimulated hepatocytes and it remains to be seen whether the modulation of
mTOR under basal conditions in primary tissues causes significant changes in
cellular redox-homeostasis.
There is increasing evidence that mTOR-regulated
autophagy plays a dual role in the cellular response to oxidative stress. On
the one hand autophagic pathways are compromised due to ageing and in
age-related disorders such as Alzheimer's and Huntington's Disease, which could
lead to accumulation of oxidized proteins in aged cells under normal growth
conditions [87,88]. This
suggests that upregulation of autophagy protects against free radical damage.
Indeed, pharmacological induction of autophagy decreased the age-dependent
accumulation of oxidatively-damaged mitochondrial DNA in rat liver [89,90]. This
view is also supported by genetic studies: for example, enhanced Atg-8
expression in old Drosophila brains extends adult life span, promotes
resistance to oxidative stress and reduces the accumulation of oxidized
proteins [55].
Additionally, the age-dependent reduction in autophagy may cause the build-up
of severely damaged mitochondria, further increasing oxidative stress and
causing additional molecular, cellular and tissue damage with age. Hence
autophagy may provide the front line of defense against oxidative stress. The
recent finding that Atg4, an essential protease that controls the lipid
modification of Atg8 and autophagosome formation, is a direct target for
oxidation by hydrogen peroxide, further underlines this idea. It has also been
suggested that low levels of ROS provide a signal to regulate autophagic
survival and death processes (reviewed in [91]).
Collectively, the evidence suggests that free radicals are upstream and
downstream of mTOR and numerous feedback and feed forward loops exist. For a
conceptual view on the role of ROS versus TOR in ageing the reader is referred
to the recent review by Blagosklonny [34].
On the other hand, in addition to its
role in promoting cell survival and increasing life span, autophagy can
actually result in cell death (autophagic or type II cell death) [92] that is
observed under conditions of oxidative stress. It has been demonstrated that
hydrogen peroxide induces autophagy via a novel autophagy signaling mechanism
that links PARP-1 activation to the LKB-1-AMPK-mTOR pathway. Hence PARP-1
activation appears to promote autophagy. Poly(ADP-ribose) polymerases are well
known for repairing single and double DNA strand breaks and therefore they play
a role in maintaining genomic stability, preventing carcinogenesis and ageing
(reviewed in [93]). In this
context it is important to note that p53, the "guardian of the cellular genome" that senses cellular damage, both
positively [94] and
negatively [95] regulates
autophagy. While nuclear p53 transactivates autophagy-enhancing genes [94,96], the
cytoplasmic pool of p53 can act as a negative regulator of autophagy. Knockout
of p53 stimulates autophagy in human, mouse and C. elegans cells [95]. The tumor
suppressor p53 favors organismal ageing [97,98]. p53
gain-of-function mutations are linked to accelerated ageing and premature death
in mice [99] and humans [100], whereas a
dominant negative p53 transgene increases longevity in Drosophila [101] and loss
of function mutations of the C. elegans p53 ortholog cep1 extends
life span [102]. Hence, a
recent study tested whether a mutation in cep1 increases life span in
worms via an increase in baseline autophagy. Tavernarakis and colleagues found
that RNAi against the autophagy gene bec-1 significantly reduced life
span extension caused by cep-1 mutants [103]. It is
likely that this functional link between increased life span and increased
autophagy due to the cep-1 mutation are TOR dependent as in mammalian
cells knockout and knockdown of p53 causes an inhibition of mTOR [95]. The exact
molecular mechanisms and signaling between p53 and (m)TOR activity and its
relationship to ageing is expected to be rather complex and remains to be
determined.
Funding in the Wyttenbach lab is provided
by the Medical Research Council (MRC), Biotechnology and Biological Sciences
Research Council (BBSRC), and the Gerald Kerkut Trust. The Proud lab is funded by AstraZeneca, the
Wellcome Trust and the British Heart Foundation. We thank Aviva M. Tolkovsky and Joel Parker for
critical comments on this review.
The authors of this manuscript have no conflict of
interests to declare.