Introduction
Cells are continuously exposed to a wide variety of
physical and chemical stresses such as oxidation, radiation and heavy metals,
which cause damage to cellular proteins, lipids and DNA. Organisms have evolved
multiple protective mechanisms to counteract these endogenous and exogenous
damages. Nevertheless, the effectiveness of these protective pathways seems to
decline with age. As such, aging can be defined as the decrease in the
probability of successful repair of cellular damage.
One of the major sources of cellular
insults is damage to DNA. To counteract detrimental DNA damage, cells are
endowed with a complex network of DNA damage response (DDR) proteins which are
capable of detecting DNA damage, and then triggering and amplifying a signaling
cascade, which ultimately leads to either cell-cycle arrest and DNA repair, or
to apoptotic cell death to eliminate permanently damaged cells [1]. The importance
of the DDR in maintaining genomic integrity and limiting the effects of aging
is highlighted by premature aging phenotype
of mice that lack key DNA repair factors [2]. More
tellingly, almost all genetic conditions that lead to premature aging in humans
have been mapped to genes belonging to the DDR [3]. Mutations
in the Werner DNA helicase, which is required for DNA replication and at
telomeres, lead to Werner syndrome and the components of the nucleotide
excision repair (NER) XPC, ERCC6, ERCC8 and the ERCC1/XPF complex involved in
inter-strand DNA crosslink repair are mutated in Cockayne Syndrome and in
Trichothiodystrophy (TTD), two prominent premature aging disorders [4]. These
findings suggest a prominent, and causal, role for DNA damage responses in
aging.
The DDR, like all major nuclear processes such as DNA
replication and transcription, operates in the context of the chromatin fiber [5,6]. Chromatin
is made up of nucleosomes, repetitive units of 146bp of DNA tightly wrapped
around an octameric core of histone proteins (H2A, H2B, H3 and H4). Nucleosomes
are further packaged into higher order structures by the action of
architectural chromatin proteins such as histone H1 and heterochromatin protein
HP1. Based on cytological criteria, chromatin is classified into euchromatin,
which is loosely packed and generally
transcriptionally active, and into heterochromatin, which is more compacted and
generally represents a transcriptionally repressive environment. Nucleosomal
histones are modified by complex patterns of post-translational modifications
(PTM) such as acetylation, methylation and ubiquitination which appear to
dictate the dynamic recruitment of non-histone proteins to chromatin and
regulate its function [7]. Furthermore, chromatin structure and function is
also determined by the methylation status of DNA itself and by a large number
of ATP-dependent remodeling factors. Both the level of chromatin compaction,
and hence the accessibility of DNA, and the recruitment of chromatin-associated
factors determine the outcome of transcription, DNA replication and DNA damage
repair. All these modifications to chromatin structure, and thus its
informational content, are inherited through several cycles of cell division
and as such represent an epigenetic memory [8].
Chromatin defects in aging
Chromatin defects are associated with aging. The first
hints pointing to a possible link between chromatin maintenance and aging came
from studies in the yeast S. cerevesiae, where the NADH-dependent Sir2
histone deacetylase Sir2 was found to be important for establishing
heterochromatin at telomeres, at ribosomal DNA (rDNA), and at HMR and HMR loci,
which encode factors needed for yeast mating type switching [9-14]. Upon
prolonged growth, equated to aging in yeast, repetitive rDNA tends to
hyper-recombine and form extrachromosomal rDNA circles (ERC), indicative of
increased chromatin fragility [15]. Formation
of heterochromatin at rDNA sites by overexpression of Sir2 reduces this
hyper-recombination and prolongs lifespan, suggesting a contribution of
chromatin structure to aging [16]. Further
experiments in worms and flies demonstrated a similar role in lifespan
extension for Sirt1, the closest orthologue of yeast Sir2 in these organisms [17,18]. Nevertheless,
the role of Sirt1 in increased longevity in higher eukaryotes might not just
involve heterochromatin maintenance, since in this case the molecular mechanism
does not seem to involve ERC stabilization [19].
Furthermore, the analysis is complicated by the fact that in mammals SIRT1
deacetylates a wide variety of non-histone, aging-related transcription factors
such as p53, HSF1 and members of the FOXO transcription factors family [20-22].
Identification of the mechanisms of action of SIRT1 in higher organisms will be
key to clarifying its role in the aging process.
There are several
other clear indications for a role of chromatin and its maintenance in aging. A hallmark of
cellular aging is the appearance of characteristic changes in the epigenetic
make-up of the genome. Epigenetic changes associated with aging in mammalian
cells include loss of DNA methylation at repetitive DNA sequences [23-25], which
are generally heterochromatinized, and an increase in DNA methylation at CpG
islands in the promoters of specific genes [26,27]. Cells
from aged individuals and patients with the premature aging disorder
Hutchinson-Gilford Progeria Syndrome (HGPS) are also characterized by loss of
heterochromatin, by loss of key architectural chromatin proteins such as HP1
and the histone mehtyltransferase EZH2, and, importantly, by alterations in the
levels of heterochromatin-associated histone PTM including H3K9me3 and H3K27me3
[28-31]. Interestingly, both prematurely and normally aged cells exhibit dramatically
increased levels of unrepaired DNA damage [30,32].
In addition to epigenetic and structural chromatin
defects, there are indications that aging in mammals is accompanied by
stochastic deregulation of gene expression. Transcriptional noise at the
single cell level increases with age in the mouse heart, most likely as a
consequence of oxidative DNA damage [33].
Furthermore, in mammalian cells oxidative DNA damage also seems to relocalize
SIRT1 from otherwise transcriptionally repressed genes to sites of DNA damage [34]. This has
led to the speculation that, through unknown mechanisms, aging disrupts the
epigenetic organization of heterochromatin both at a global and at a
gene-specific level, thus leading to elevation of stochastic transcriptional
noise and to the disruption of transcriptional programs necessary for proper
cell homeostasis [35]. In
contrast to this model of stochastically occurring defects in gene expression
programs, the aging process seems to also induce a specific transcriptional
response, which dampens the somatotrophic IGF-1 axis and helps protecting cells
from DNA damage and stress [36].
The study of chromatin in aging also points to a key
influence of aberrant chromatin structure on aging-related defects in DNA
repair. Impairment of SIRT1 leads to defective DNA damage repair in mammalian
cells [34] and a knock-out mouse model for SIRT6 shows signs of premature aging
and has defects in the base excision repair pathway [37]. The exact
molecular basis for these phenotypes is not clear yet. One possibility is that
SIRT6 affects genomic stability by regulating the levels of H3K56Ac [38,39], a PTM
important for chromatin assembly and DNA damage tolerance in yeast [40,41].
A molecular mechanism for aging-associated chromatin
defects
The molecular mechanisms leading to
chromatin defects in aging are largely unknown. Recent analysis of chromatin
defects in the premature aging disease HGPS have given some of the first
insights into how chromatin ages [42]. HGPS is an
extremely rare genetic disease caused by a de novo point mutation in the
lamin A (LMNA) gene, a major structural component of the nuclear envelope [43].The
pathogenic mutation leads to the production of an internally truncated form of
lamin A, referred to as progerin. This protein acts in a dominant-negative gain
of function fashion causing the diverse and pronounced chromatin defects.
Analysis of the molecular mechanisms involved in bringing about chromatin
defects in HGPS and old cells uncovered the NURD complex as a key player in
aging [42]. NURD is a
ubiquitous chromatin remodeling complex which contains the histone deacetylases
HDAC1 and HDAC2 and the ATPases CHD3 and CHD4 as catalytic subunits. NURD has
been implicated in transcriptional repression at specific promoters and more
recently has also been shown to associate with pericentromeric heterochromatin [44,45]. The
protein levels and the activity of several NURD components including HDAC1 and
the histone chaperones RBBP4/, are reduced in HGPS cells and normally aged
cells. A direct role for NURD loss in aging-associated chromatin defects is
indicated by the finding that knock-down of NURD members in normal cells
recapitulates aging-related chromatin defects including heterochromatin loss
and increased DNA damage [42]. NURD is
known to be involved in a variety of chromatin functions and its loss may
explain the broad spectrum of chromatin defects seen in aged cells [42].
Chromatin structure as a trigger of aging
There is little doubt that chromatin
defects and DNA damage play a part in the aging process. The unresolved question
is: how? One recently proposed scenario suggests that DNA damage and the
cellular response to it leads to chromatin defects via relocation of epigenetic
machinery from its normal distribution in the genome and to structural chromatin
changes, eventually resulting in gene misregulation [34] (Figure 1A). An
alternative possibility is that the aging process is triggered by loss of
chromatin structure, leading to altered epigenetic modifications, and increased
susceptibility to DNA damage. In this model DNA damage is a downstream event (Figure 1B). The
key question to distinguish between these two models is: what comes first, DNA
damage or chromatin defects? A partial answer comes from recent observations in
the premature aging disorder HGPS. Upon induction of the dominant negative
disease-causing protein in normal skin fibroblasts, chromatin defects occurred
prior to DNA damage [42][41]. Further support for a trigger role of chromatin
structure in DNA damage and aging, is the observation that suppression of the
activity of chromatin modifiers generates high levels of endogenous DNA damage,
as seen in the case of several subunits of the NURD complex [42], the SET8
H4K20 histone methylase [46,47], and
for the Su(var)3-9 H3K9 histone methylase in Drosophila [48]. In these
cases chromatin structural defects clearly precede DNA damage, placing
epigenetic and chroma-tin structure changes upstream of DNA damage events.
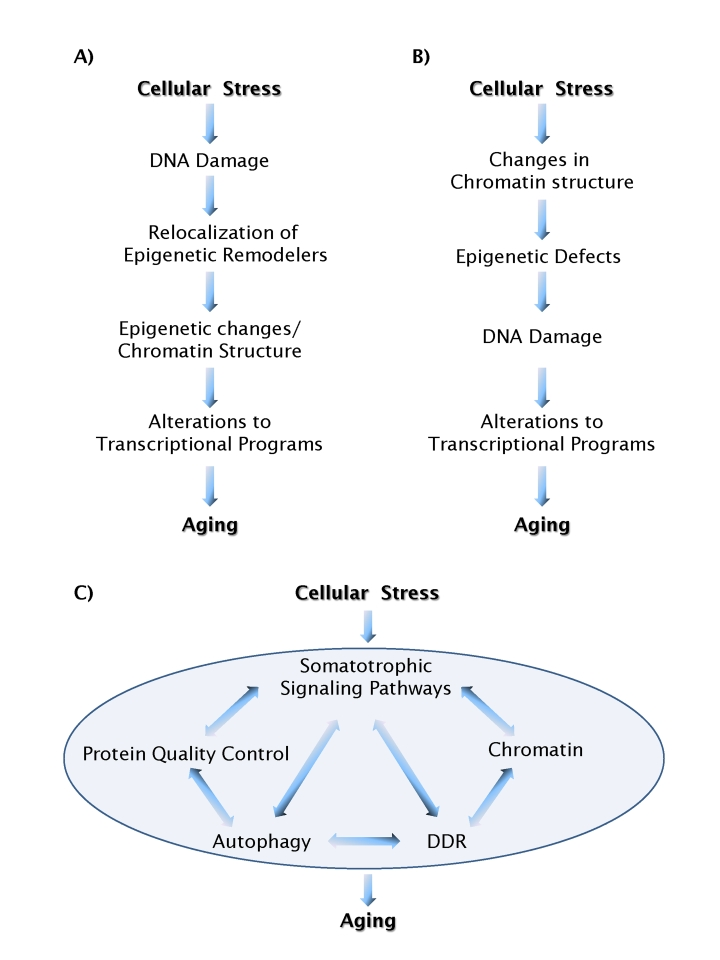
Figure 1. Models of aging pathways. (A) A scenario in which DNA
damage acts as a causal trigger for aging. (B) A scenario in which
chromatin structure acts as a causal trigger for aging. Feedback loops,
which are likely to exist between most individual events, are not shown for
simplicity. (C) Chromatin structure and DNA damage pathways act in
an integrated fashion with a multitude of other cellular process to form a
network of aging processes.
How may aberrant chromatin structure lead to DNA
damage and aging? Although only poorly investigated and understood, it is
becoming clear that chromatin structure affects the susceptibility of DNA to
damage and progression of the DDR [5]. DNA repair
occurs with slower kinetics in highly condensed heterochromatin, presumably
due to the inability of repair factors to rapidly access the site of damage [49].
Furthermore, heterochromatinized regions of the genome, like nucleoli,
centromeres and telomeres tend to be rich in repetitive sequences that are
particularly prone to recombination. As such it is possible that the compacted
nature of heterochromatin suppresses hyper-recombination of repetitive
sequences, the formation of aberrant DNA structures and genomic instability [50]. Another,
not-mutually exclusive, possibility is that altered chromatin structure
increases the steady-state level of DNA damage due to replication defects such
as impaired passage of the replication machinery or to replication fork
stalling. It is indeed possible that intact heterochromatin conformation is
necessary for the DNA replication machinery to properly proceed through highly
repetitive portions of the genome. This last hypothesis is in line with the
observation that siRNA silencing of either the histone-chaperones RBBP4/7 [42] or of SET8 [46,47] impairs
S-phase progression.
Although these observations point towards an upstream
role of chromatin structure in determining DNA stability, it is also true that
genome integrity influences chromatin structure. Local DNA damage affects the
epigenetic status of chromatin both in the vicinity of a lesion through phosphorylation,
acetylation and ubiquitination of nearby histones, but also globally [51]. In response
to local DNA damage, the zinc finger protein KAP1 is phosporylated by the ATM
kinase and released from heterochromatin, thus facilitating the access of DNA
repair factors to these more compacted regions of the genome and also
potentially altering chromatin structure at other sites [51].
Furthermore, as previously mentioned, DNA damage results in redistribution of
chromatin associated factors and histone modifiers like SIRT1, possibly leading
to profound changes in the transcriptional regulation of genes [34]. Clearly,
the relationship between chromatin structure and DNA damage is not
unidirectional, but rather a mutual one.
A network of aging mechanisms
In our search of molecular mechanisms for
biological processes we usually look for linear pathways. What we are learning
about the interplay between chromatin structure, epigenetic regulation and DNA
repair makes it clear that this is not a one-way street and that these
processes are likely connected and linked by feedback mechanisms. The most
likely scenario is that chromatin structure, epigenetic status, and DNA repair
represent nodes of a network of processes involved in protecting cells from
endogenous and exogenous insults, ultimately leading to increased longevity (Figure 1C).
Importantly, these processes do not work in isolation, but instead are linked
to pathways dedicated to maintain proteostasis such as the heat shock response
or autophagy, and hormonal regulation of cellular growth, with the mTOR and
IGF-1 pathways, whose role in the regulation of longevity has already been
established in mammals [52-54]. In
support of a branched network of cellular functions involved in aging, possible
connections between the DNA damage, the inhibition of the IGF-I and mTOR
pathways have been suggested [55]. This
scenario is supported by observations in cells from the ZMPSTE24-/- mouse, a
murine model of HGPS in which lamin A processing is impaired. The progeriod
ZEMPSTE24-/- mouse shows dramatic alterations in heterochromatin architecture,
accompanied by increased DNA damage, by the activation of the authophagic
response and by downregulation of the mTOR pathway [32,56].
Aging is a complex process. It is hardly realistic for
it to be explained by a single pathway or even a set of closely related
pathways. More likely, many diverse cellular functions will contribute to aging
and they will do so in a highly inter-dependent manner. The recent
investigation of the role of chromatin structure, epigenetic modifications and
DNA damage in aging makes this clear. While we are still struggling to
understand the precise relationship of these events in the aging process, we
are already discovering links to more distantly related events such as
signaling pathways and metabolism. Rather than attempting to explain aging as
the consequence of degeneration of single pathways, a conceptual framework
consisting of a network of affected processes not only reconciles different, at
times contentious hypotheses regarding aging mechanisms, but will ultimately
lead to an integrated view of these processes and to a more accurate
understanding of the molecular basis of aging.
The authors of this
manuscript have no conflict of interest to declare.