Introduction
Caloric restriction (CR) reduces the
levels of multiple aspects of inflammation [1-3], suggesting a link between
energy status and inflammation. This linkage is enforced by recent progress in
obesity research. Chronic inflammation is widely observed in obesity (metabolic
syndrome). The obesity-associated inflammation is involved in pathogenesis of
type 2 diabetes, hypertension, atherosclerosis, fatty liver, cancer metastasis,
and asthma in obesity. Obesity has a higher prevalence in the aging population
as a result of reduced energy expenditure with less physical activity. Physical
activities consume a major portion of energy in our daily life, which are
usually reduced in the aging population. This reduction in energy expenditure
may lead to energy accumulation in the body and consequently a gain in
adiposity. In obesity, systemic chronic inflammation occurs with elevated proinflammatory
cytokines (IL-6, MCP-1, CRP, PAI-1,
et al.) in the circulation. The systemic inflammation
is due to an inflammatory response in adipose tissues that are under quick
expansion. Adipocytes produce these cytokines. In addition, macrophage
infiltration into the adipose tissue contributes significantly to the cytokine
production. Although we have learned a lot about the signaling pathways that
link energy accumulation (adiposity) to chronic inflammation, we know little
about the real biological significance of the inflammation. This article
addresses this issue, and provides an overview of the interaction of
inflammation and energy balance.
1.
Chronic inflammation from energy accumulation
In
obesity research, the link between chronic inflammation and energy (fat)
accumulation is well established. The initial observation of TNF-α elevation in adipose tissue of obese mice provides
the first evidence for the chronic inflammation in 1993 by Hotamisligil and
colleagues [4]. Thereafter, the concept was enforced by abundant literature
identifying increases in many other inflammatory cytokines, such as plasma
C-reactive protein (CRP), interleukin 6 (IL-6), plasminogen activator
inhibitor-1 (PAI-1), in models of obesity. Activation of inflammatory kinases
such as IKKβ (IkBα kinase beta) and JNK1 (c-Jun N-terminal kinase 1)
provides additional evidence for activation of intracellular inflammatory
pathways in obesity [5-6]. Obesity-associated inflammation is chronic,
systemic, low-grade, and not linked to any infection. In contrast to
inflammation induced by bacteria or virus infection where neutrophil
granulocytes are elevated in the circulation, neutrophil granulocytes are not
increased in blood in obesity. The inflammation is systemic since the
inflammatory cytokines are increased in the circulation. The inflammation is at
a low grade in obesity since there is no fever and malaise, which are often
observed for inflammation associated with bacteria/viral infection.
2.
Inflammation origin: Energy accumulation may induce inflammation through
metabolites of fatty acids and glucose (Figure 1)
The
metabolites of fatty acids and glucose include diaglyceride (DAG), Ceramide,
and reactive oxygen species (Figure 1). They activate inflammatory response
through several approaches. They may direct interact with signaling kinases
(PKCs, JNKs and IKKs) in cells [7]. They may also act through cell membrane
receptors for lipids, such as TLR4, CD36 or GPR [8-11]. The reactive oxygen
species (ROS) are generated from fat or glucose oxidation in mitochondria. ROS
may induce activation of the inflammatory kinases (JNK and IKK). The lipids
also induce endoplasmic reticulum (ER) stress for activation of JNK and IKK
[12-13]. In CR, these metabolites of glucose and fatty acids are reduced from
less calorie intake. The risk of inflammation is reduced.
In obesity, adipose tissue is a major
source of chronic inflammation [14-15]. In adipose tissue, adipocytes and
adipose tissue macrophages (ATM) are the major cell types responsible for the
production of inflammatory cytokines. The representative cytokines include TNF-α, IL-6, MCP-1 and PAI-1. Adipokines (Leptin and adiponectin) are
produced by adipocytes and also involved in the regulation of inflammation.
Macro-phages and adipocytes are activated during the process of adipose tissue
expansion. Recent studies suggest that the adipose tissue expansion induces a
local hypoxia response [16]. The hypoxia response serves as a common root for
all of the stress responses in adipose tissue, such as oxidative stress, ER
stress, and inflammatory stress [17-19]. Hypoxia directly promotes the chronic
inflammation through activation of transcription factors (NF-kB and HIF-1) in
adipocytes and macrophages [16]. The hypoxia response is a result of tissue
expansion. In CR, adipose tissue expansion is reduced or under controlled. The
risk factors for inflammation, such as adipose tissue hypoxia, lipid
accumulation, ER stress and oxidative stress are all reduced or absent. These
may explain why CR reduces the risk for chronic inflammation in the body.
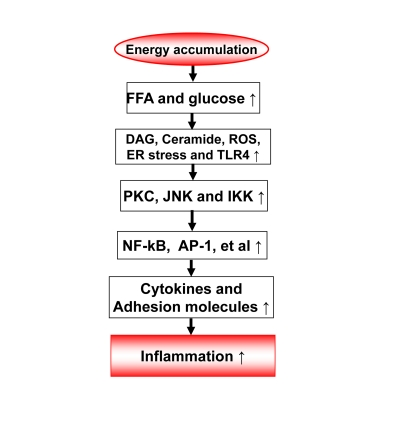
Figure 1. Energy accumulation induces inflammation. Energy accumulation leads to elevation
in glucose and fatty acids. These substrates lead to production of
diaglycerids (DAG), Ceramide, reactive oxygen species (ROS) and activation
of toll-like receptor 4 (TLR4) in cells including macrophages and endothelial
cells. All of these events may activate the inflammatory signaling
pathways, such as IKK/NF-kB and JNK/AP-1. As a consequence, expression of
inflammatory cytokines and adhesion molecules may increase for chronic
local inflammation. When inflammatory cytokines are elevated in the
circulation, the energy accumulation causes systemic chronic inflammation,
which is observed in obesity. This kind of chronic inflammation is limited
or prevented by calorie restriction
3.
Inflammation feedback to energy accumulation
The
inflammation observed in adipose tissue likely serves as a feedback signal
locally in adipose tissue and systemically for energy expenditure (Figure 2).
In adipose tissue, inflammation inhibits adipocyte expansion and adipocyte
differentiation, changes adipocyte endocrine and induces extracellular matrix
remodeling [20]. The local response is translated into a systemic response
through cytokines and free acids released from adipose tissue.
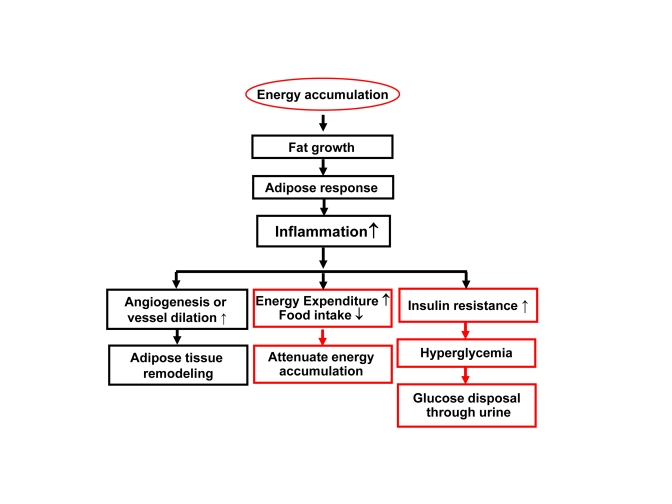
Figure 2. Inflammation in obesity. Rapid growth of
adipose tissue leads to quick expansion of adipose tissue. When
angiogenesis or vessel dilation can not meet the demand for blood supply,
there will be an adipose tissue hypoxia (ATH) from lack of blood supply.
ATH will induce angiogenesis and trigger inflammation. Inflammation will
promote angiogenesis and vasodilation locally in the tissue for
extracellular remodeling. When inflammatory cytokines and fatty acids are
elevated in the circulation, they will promote energy expenditure
systemically. The inflammatory response may also induce hyperglycemia and
energy disposal through glucose excretion in urine. In this way,
inflammation acts through insulin resistance and hyperglycemia.
(a) Adipocyte
inhibition. A major function of adipocytes is to store fat. In addition, the
adipocytes secrete many cytokines/hormones in its endocrine activity. Inflammatory
cytokines inhibit adipocyte function in multiple aspects. These include
inhibition of preadipocyte differentiation, induction of lipolysis and
suppression of adiponectin expression in mature adipocytes. These inhibitory
activities are well documented for TNF-α and IL-1 [21-23]. At the molecular level, inflammation
inhibits insulin signaling pathway [24-26] and PPARγ activities in adipocytes [27].
These effects contribute to suppression of tissue expansion, and alteration in
cytokine profile. The disorders in lipid metabolism and cytokine balance
contribute to the whole body insulin resistance, a result of impaired insulin
signaling in multiple organs (skeletal muscle, liver, and adipose tissue)
[28-30]. Insulin resistance may induce hyperglycemia, which in turn leads to
glucose excretion through urine (type 2 diabetes). The type 2 diabetes is an
extreme condition in the body to get ride of energy surplus in an effort to
prevent energy accumulation in the body.
(b) Adipose tissue remodeling: Macrophage infiltration
is a major marker of local inflammation in the adipose tissue in obesity.
Adipose tissue macrophages (ATM) have been under active investigation since
2004 when macrophage infiltration was initially identified in obese mice
[31-34]. The discovery provides a source for TNF-α
in adipose tissue since mature adipocytes produces very little TNF-α [31-34]. The biological significance of macrophage
infiltration remains to be elucidated. However, more and more evidence suggests
that macrophages are required for adipose tissue remodeling and adipogenesis of
preadipocytes. Macrophages may serve as a signal amplifier in the adipose
tissue for stimulation of angiogenesis [35]. Macrophages produce many
angiogenic factors, such as PDGF, TGF-β and HGF, which are
increased in adipose tissue in obese individuals [36-37]. Interestingly, this
activity of macrophages is required for adipose tissue growth in lean mice
[38-39] and obese mice [35]. Macrophages may also regulate blood flow through
production of vasodilators (such as NO). Macrophages may clean the cell debris
of dead adipocytes within the adipose tissue [40]. An increase in adipocyte
death was reported in the adipose tissue of obese mice, and the dead cells were
surrounded by ATMs to form the "Crown" like structure [40-41]. The cell death
in adipose tissue may be a result of the hypoxia response [42]. In CR, the
adipose tissue expansion is under control, there are not such risk factors for
macrophage activation in adipose tissue.
(c) Fuel mobilization. Inflammation regulates fuel
mobilization. Fuel (fatty acids) mobilization from adipose tissue to other
tissues is controlled by the nervous system and hormones/cytokines. The role of
inflammatory cytokines has drawn a lot of attention in the fuel mobilization.
Cytokines such as TNF-α, IL-1, IL-6, et al., activate fuel efflux in
adipocytes through lipolysis, in which free fatty acids (FFAs) are generated
from triglycerides under hydrolysis and released into blood stream. FFAs are
normally oxidized in mitochondria for ATP production. An increase in FFA supply
may lead to acceleration of energy expenditure. However, when FFA supply
overrides the consumption, they deposit in non-adipocytes in the form of
ectopic fat deposition. The ectopic fat contributes to pathogenesis of fatty
liver disease and atherosclosis (deposit on the blood vessel wall). In the
physiological conditions, IL-6 secreted by contracting muscle is involved in
coordination of fuel mobilization between adipose tissue and skeletal muscle
during exercise [43-44]. In CR, the fatty acid supply is limited as a result of
reduced calorie intake, the risk for ectopic fat deposition will be reduced.
This may help in prevention of fatty liver and atherosclosis.
(d) Energy intake. Inflammatory cytokines are involved
in the regulation of energy intake and expenditure. IL-1 and IL-6 reduces food
intake and prevent hyperphagia [45-46]. Cytokines (IL-1, IL-6 and TNF-α)
also induce energy expenditure [46-50]. These activities of cytokines are
dependent on their actions in the central nervous system [46-47,51-52].
Therefore, inflammatory cytokines may serve as an anti-obesity signal by
modifying both energy intake and energy expenditure. Additionally, these data
indicate that the inflammatory cytokines may serve as a link between peripheral
tissues and central nervous system in the control of energy balance.
4. Energy expenditure by inflammation
The activities of inflammatory cytokines
on adipocytes and neurons suggest that inflammation may inhibit energy
accumulation. They induce energy expenditure and inhibits food intake. These
possibilities are strongly supported by phenotypes of transgenic mice with
chronic inflammation and by cytokine infusion studies. Transgenic mice of
IKK2/NF-kB have provided new evidence.
The
IKK2/NF-kB pathway is a dominant inflammation signaling pathway. The pathway
has been under active investigation in the obesity field after IKKβ was
found to induce insulin resistance in obese mice [5]. The serine kinase IKK has
three major isoforms including IKKα (IKK1), IKKβ (IKK2) and
IKKγ, in which IKKβ is required for NF-kB activation [53]. In obesity,
IKKβ is activated by several intracellular signals, such as ROS, ER
stress, DAG, and Ceramide. IKKβ is also activated by the extracellular
stimuli including TNF-α, IL-1, and fatty acids [8], and hypoxia [54].
IKKβ induces NF-kB activation by phosphorylation of the Inhibitor Kappa B
alpha (IkBα) [55].
NF-kB
(nuclear factor kappa B) is a ubiquitous transcription factor that is formed by
two subunits of Rel family, which include seven members, p65 (RelA), p50
(NF-kB1), c-Rel, RelB, p100, p105, p52 [56]. These members form a homodimer or
heterodimer in the regulation of gene transcription. In most case, NF-kB is a
heterodimer of p65 and p50. P65 contains the transactivation domain and
mediates the transcriptional activity of NF-kB. P50 usually inhibits the
transcriptional activity of p65 [57], and the inhibition disappears in the
NF-kB p50 knockout mice [58]. In the classical pathway, NF-kB activation is
mediated by IKKβ-induced phosphorylation, proteasome-mediated degradation
of IkBα [53]. In response to stress responses, NF-kB promotes
lipid mobilization through suppression of PPARγ activity in
the nucleus [59]. It also induces transcription of inflammatory cytokines (TNF-α,
IL-1, IL-6, MCP-1, et al.). In the alternative pathway, NF-kB is activated by
hypoxia in the absence of IkBα degradation. This type of NF-kB
activation in adipocytes and macrophages contributes to chronic inflammation in
the adipose tissue of obese individuals [16].
NF-kB
activity may promote energy expenditure. This activity of NF-kB is supported by
documents on energy expenditure in cachexia [60-61] and infection. However, the
role of NF-kB in energy expenditure was not tested in transgenic models. To
this point, we investigated energy metabolism in transgenic mice with elevated
NF-kB activities. The transcriptional activity of NF-kB is enhanced either by
over-expression of NF-kB p65 (RelA) in the fat tissue, or inactivation of NF-kB
p50 (NF-kB1) by global gene knockout [65]. In these two models, inflammatory
cytokines (TNF-α and IL-6) were elevated in blood and energy expenditure
was increased in day and night [65]. The oxygen consumption and CO2 production
were both increased in the mice. Locomotion
was not altered, but food intake was increased in the mice. Expression of
inflammatory cytokines (TNF-α and IL-6) was elevated in adipose tissue and
macrophages. On a high fat diet (HFD), both lines of transgenic mice were
protected from obesity and insulin resistance [65-66]. The data suggests that
the transcription factor NF-kB promotes energy expenditure and inhibits energy
accumulation. The inflammatory cytokines may mediate the NF-kB activity in
energy expenditure. In the mice, lipid accumulation is prevented by the
enhanced energy expenditure. The studies suggest that inflammation may prevent
insulin resistance by eliminating lipid accumulation. IKKβ was investigated in the control
of insulin sensitivity [5,62-63] and food intake in transgenic mice [64].
However, IKKβ was not
investigated in the control of energy expenditure in these studies.
NF-kB
may promote energy expenditure through the inflammatory cytokines. In the two
transgenic models, systemic inflammation was observed with elevated proteins
for TNF-α and IL-6 in the serum [65-66]. Expression of TNF-α and IL-1
mRNA was increased in adipose tissue and macrophages. These cytokines are
positively associated with energy expenditure in the body [61]. In transgenic
mice with deficiency in these cytokines or their receptors, energy accumulation
is enhanced, suggesting a reduction in energy expenditure. This positive energy
balance was reported in transgenic mice with deficiency in TNF-α [50],
IL-1 [45] or IL-6 [46]. On the other side, when these cytokine activities are
enhanced in transgenic mice, energy accumulation is decreased leading to a lean
phenotype [48-49,67-68]. The cytokines may act in the hypothalamus of central
nervous system to regulate the energy balance [46-47,51-52]. In addition to
the central mechanism, activation of mitochondria by the cytokines in the
peripheral tissues may also contribute to the energy expenditure. TNF-α
and IL-1 enhances mitochondrial function through phosphorylation-mediated
activation of PGC-1α [69]. This activity of inflammatory cytokines may
contribute to energy consumption in mitochondria-enriched tissues/organs such
as liver, skeletal muscle and brown fat. Inflammation may be a drug target in
the management of energy metabolism [70-71].
5.
CR and chronic inflammation
Studies have demonstrated that CR
decreases the circulating levels of inflammatory cytokines and inflammatory
signaling activities in a wide variety of tissues [1-3]. CR is able to decrease
global levels of inflammatory responses in the body. Interestingly, the
beneficial effects of CR may be related to a decrease in visceral fat and
adipose reactivity [3,72]. It has been documented that adiposity during aging
contributes to a number of morbidity factors including insulin resistance,
dyslipidemia, atherosclerosis, hypercoagula-bility and hypertension [73-74]. However,
it is important to remember that the most inflammation data are derived from
the visceral fat and ectopic fat [72-74]. For example, subcutaneous fat has
been observed to have beneficial effects on lipid and energy homeostasis, and
even counteract the negative effects of visceral adipose tissue [75]. It is
important to note that CR has beneficial effects in non-obese humans as well as
non-obese rodents [76-77], indicating that decreased adiposity may not be the
only mediator of beneficial effects of CR. This fact suggests that a decrease
in energy accumulation is more important in the control of inflammation since
this may apply to both obese and non-obese conditions.
This
study is supported by NIH fund (R56DK068036-6) and ADA research award
(7-07-RA-189) to Ye J.
The authors of this manuscript have no conflict of
interests to declare.