An animal model manifesting neurodegeneration and obesity
Abstract
Although the existence of a link between neurodegenerative diseases and obesity has been suggested, a causal relation between neural degeneration and obesity has remained to be demonstrated experimentally. We recently showed that neurodegeneration in the hypothalamic satiety center results in obesity in mice transgenic for E4B (also known as UFD2a), a mammalian ubiquitin elongation factor (E4). Increased expression of E4B in neurons of the transgenic mice results in the formation of ubiquitin-positive aggregates similar to those apparent in many human neurodegenerative diseases as well as in degeneration of hypothalamic neurons responsible for the regulation of food intake and energy expenditure. We thus propose that neurodegeneration is a possible cause of human obesity and related metabolic diseases, which have become a serious public health problem worldwide. Our animal model is thus a powerful tool for studies of the relation between neurodegeneration and obesity.
Aging of the human population is a key
concern worldwide because of the associated social and medical problems.
Important diseases related to aging include neurodegenerative conditions, such
as Alzheimer's disease, most of which are characterized by the formation of
intracellular protein aggregates in neurons and neuronal loss. Individuals with
such diseases exhibit various neural disorders including motor, cognitive, and
behavioral dysfunction. Another disease that has traditionally been associated
with aging is obesity, although this condition, together with its accompanying
metabolic abnormalities, has recently also begun to affect younger individuals
as a result of changes in diet and lifestyle and has become a serious public
health problem worldwide. A link between these two types of disease has been
postulated on the basis of their association with aging. Indeed, the possible
relation between neurodegeneration and obesity in animal models or humans has
been studied now for several decades. However, most such studies have focused
on the possibility that obesity and related metabolic disorders exacerbate
neurodegeneration and
thereby promote cognitive decline and increase
vulnerability to brain injury [1]. Few studies have addressed the possibility
that neurodegeneration in the brain may cause obesity, as is suggested by the
identification of hereditary neurodegenerative disorders associated with
obesity such as Prader-Willi syndrome [2].
E4
as a new player in the ubiquitin-proteasome system
A key focus of our research group has been the
functions and underlying mechanisms of the ubiquitin-proteasome system (UPS).
The UPS plays an important role in the elimination of short-lived regulatory
proteins [3], including those that contribute to such processes as the cell
cycle, cellular signaling in response to environmental stress or extracellular
ligands, morphogenesis, secretion, DNA repair, and organelle biogenesis [3-5].
The UPS pathway includes two key steps: covalent attachment of multiple
ubiquitin molecules to the protein substrate and degradation of the
ubiquitylated protein by the 26S proteasome complex. The system responsible for
the attachment of ubiquitin to the target protein consists of several
components that act in concert [3,6], including a ubiquitin-activating enzyme
(E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin-protein isopeptide
ligase (E3). E3 is thought to be the component of the ubiquitin conjugation
system that is most directly responsible for substrate recognition. In
addition, a new type of ubiquitylation enzyme, a ubiquitin chain assembly
factor (E4), was recently discovered and shown to be required for the
degradation of certain types of substrate, including an artificial fusion
protein with an NH2-terminal ubiquitin moiety, via a ubiquitin fusion
degradation (UFD) pathway [7,8]. Ufd2 of Saccharomyces cerevisiae is the
prototype E4 enzyme. Ufd2 contains a conserved U-box domain, which appears to
be an essential functional domain for E4 activity [9,10], and is associated
with Cdc48 [8], which belongs to the large family of AAA-type ATPases that are
thought to possess chaperone activity [11,12]. We have previously shown that
mouse E4B (also known as UFD2a) is a homolog of yeast Ufd2, given that it
contains a conserved U-box domain at its COOH-terminus and interacts with VCP,
a mammalian ortholog of yeast Cdc48. These properties of E4B suggest that the
association of AAA-type ATPases with Ufd2-like proteins that possess
ubiquitylation activity has been conserved through evolution and may thus be
functionally important [10,13].
The roles of E4B in vivo have remained
largely unknown, however. E4B is expressed predominantly in neural tissues of
adult mice [10], suggesting that it performs a neural-specific function. We
found that E4B targets the pathological form of ataxin-3—in which abnormal
expansion of a polyglutamine tract is responsible for spinocerebellar ataxia
type 3 (SCA3) in humans—for ubiquitylation and degradation in mammalian cells
as well as in a Drosophila melanogaster model of SCA3 [14]. Furthermore, we
isolated FEZ1 (fasciculation and elongation protein zeta 1), a protein
implicated in neurite extension, as a binding partner of E4B [15]. FEZ1 is a
mammalian homolog of Caenorhabditis elegans UNC-76, which is required for
axonal bundling and elongation in the nematode [16], suggesting that a FEZ1-E4B
system also participates in axonal outgrowth and fasciculation in mammals.
Other groups also reported that UFD2a is implicated in the process of Wallerian
degeneration of neurons [17,18]. Moreover, we showed that E4B+/- mice manifest
axonal dystrophy in the nucleus gracilis as well as degeneration of Purkinje
cells associated with endoplasmic reticulum stress, and that these animals
develop a neurological disorder [13]. Mice nullizygous for E4B died in utero as
a result of developmental defects in the heart, suggesting an additional role
for E4B in developmental processes in this organ. In spite of these various
observations, however, the precise physiological functions of this enzyme
remained elusive.
Neurodegeneration and obesity in mice transgenic for
E4B
During further studies to explore the roles of E4B, we
discovered that overexpression of E4B in a neural cell line resulted in the
formation of protein aggregates that were recognized by antibodies to ubiquitin
as well as by those to p62, a marker of ubiquitin-associated aggregates. This phenomenon
was also reproduced in E4B transgenic (Tg) mouse lines in which expression of
the E4B transgene is controlled by the promoter of the gene for the mammalian
prion protein [19] (Figure 1A). This aggregate formation is apparently
dependent on the ubiquitylation activity of the enzyme, given that few such
aggregates were detected in cells expressing E4B(ΔU), a
truncated form of E4B that lacks the catalytic U-box domain (Figure 1A). In
addition, an important feature of the aggregates is that they resemble
ubiquitin- and p62-positive aggregates observed in many human neurodegenerative
diseases or in mice with neurodegeneration resulting from defects in autophagy,
another pathway for the clearance of cellular components [20,21].
The aggregate formation in E4B Tg mice was apparent
specifically in certain hypothalamic nuclei. Among these nuclei, the
aggregate-associated neurodegeneration was most obvious in the paraventricular
nucleus (PVN). PVN neurons are activated by signaling downstream of food intake
[22-25], and they function as a satiety center. Indeed, lesions in the PVN
result in the development of hyperphagic obesity in rat [26]. Furthermore,
neurodegeneration-associated gliosis was observed in the region adjacent to the
PVN in the hypothalamus of E4B Tg mice (Figure 1B), indicating that ectopic
expression of E4B results in the formation of ubiquitin-positive aggregates and
associated pathological features characteristic of neurodegenera-tive diseases
[27].
Surprisingly, the E4B Tg mice
were unequivocally obese (Figure 1C) and manifested increased lipid
accumulation in tissues such as adipose tissue and the liver [27]. We
investigated whether this obese phenotype was attributable to functional
impairment ofthe hypothalamic satiety center. The animals exhibited increased food
intake and decreased energy expenditure as well as several abnormal responses
of the center to satiety input, indicating that malfunction of the hypothalamic
satiety center is responsible for the
obese phenotype of the E4B Tg mice. Finally, we observed that the Tg mice
manifested metabolic disorders seen in obese humans.
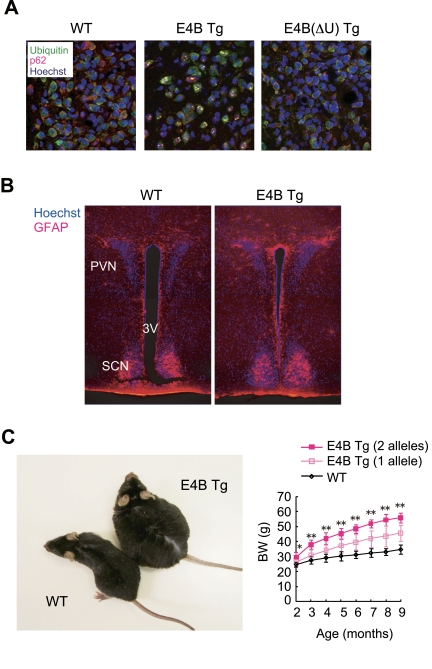
Figure 1. E4B
transgenic (Tg) mice as a new obesity model with hypothalamic
neurodegeneration. (A) Immunofluorescence analysis
of the PVN region of 6-month-old wild-type (WT) or E4B(ΔU) Tg mice and
of a 4-month-old E4B Tg mouse. Brain slices were stained with antibodies to
polyubiquitin (green) and to p62 (red), and nuclei were stained with
Hoechst 33258 (blue). Protein aggregates reacted with both types of
antibody in the PVN region of E4B Tg mice, but not in that of WT or
E4B(ΔU) Tg mice. (B) Immunofluorescence analysis of the PVN
region of 10-week-old WT or E4B Tg mice with antibodies to glial fibrillary
acidic protein (GFAP, red). Nuclei were stained with Hoechst 33258 (blue).
SCN and 3V indicate the suprachiasmatic nucleus and third ventricle,
respectively. The number of GFAP-positive glial cells in and around the PVN
was increased in E4B Tg mice, indicative of gliosis associated with
neurodegeneration. (C) Obesity in E4B Tg mice. The gross appearance
of an E4B Tg mouse and a WT littermate at 9 months of age is shown on the
left. The time course of body weight (BW) for WT mice and E4B Tg lines
harboring one or two alleles of the transgene is shown on the right. The
extent of obesity in the Tg animals harboring two alleles of the transgene
was about twice that in littermates harboring only one allele, indicating
that the obese phenotype is directly related to the expression level of the
transgene. *P < 0.05, **P < 0.01 for the Tg line with
two alleles of the transgene versus wild-type mice.
On the basis of our observations, we proposed that the
E4B Tg mouse is a new animal model for neurodegeneration-associated obesity
that possesses several advantages. First, these animals spontaneously develop
obesity and thus do not need to be fed a high-fat diet. Second, they manifest
abnormalities in the highly restricted area of the hypothalamic satiety center
and thus exhibit pathological features similar to those of some other mouse
models of obesity, such as ob/ob and db/db mice, in which the hypothalamic
leptin circuit is impaired [28,29]. Third, only one allele of the E4B
transgene is required for mice to develop obesity. Furthermore, the extent of
obesity can be varied by selection of transgenic lines with different levels of
expression or different numbers of alleles of the transgene (Figure 1C),
whereas most other mouse models are loss-of-function mutants and therefore
require homozygosity of the mutant allele for manifestation of the phenotype.
Fourth, E4B Tg mice also develop leptin and insulin resistance, glucose
intolerance, hypercholesterolemia, and hypoadipo-nectinemia during progression
of the obesity phenotype. These characteristics thus suggest that E4B Tg mice
recapitulate the course of human obesity.
Perspective
Our genetic mouse model has also provided the first
experimental demonstration that neurodegeneration can indeed result in obesity,
suggesting that some cases of human obesity might be attributable to
hypothalamic neurodegeneration in aged individuals without any other neural
disorders including cognitive and behavioral dysfunction. Aberrant activity of
E4B might be a possible cause of obesity and associated metabolic disorders in
humans, a notion that is consistent with the localization of obesity-related
genetic markers in the vicinity of the E4B gene locus [30,31]. Further analysis
of E4B function, particularly through identification of its substrates, should
provide greater insight into the pathological properties of the molecule. More
generally, nonspecific neurodegeneration associated with aging might result in
a tendency to become obese. Together, our findings with E4B Tg mice open a new
field of research linking obesity and aging processes as represented by
degeneration of neural tissue.
Conflicts of Interest
The authors of this manuscript have no
conflict of interests to declare.
References
-
1.
Bruce-Keller
AJ
, Keller
JN
and Morrison
CD.
Obesity and vulnerability of the CNS.
Biochim Biophys Acta.
2009;
1792:
395
-400.
[PubMed]
.
-
2.
Ristow
M
Neurodegenerative disorders associated with diabetes mellitus.
J Mol Med.
2004;
82:
510
-529.
[PubMed]
.
-
3.
Hershko
A
and Ciechanover
A.
The ubiquitin system.
Annu Rev Biochem.
1998;
67:
425
-479.
[PubMed]
.
-
4.
Weissman
AM
Themes and variations on ubiquitylation.
Nat Rev Mol Cell Biol.
2001;
2:
169
-178.
[PubMed]
.
-
5.
Nakayama
KI
and Nakayama
K.
Ubiquitin ligases: cell-cycle control and cancer.
Nat Rev Cancer.
2006;
6:
369
-381.
[PubMed]
.
-
6.
Scheffner
M
, Nuber
U
and Huibregtse
JM.
Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade.
Nature.
1995;
373:
81
-83.
[PubMed]
.
-
7.
Johnson
ES
, Ma
PC
, Ota
IM
and Varshavsky
A.
A proteolytic pathway that recognizes ubiquitin as a degradation signal.
J Biol Chem.
1995;
270:
17442
-17456.
[PubMed]
.
-
8.
Koegl
M
, Hoppe
T
, Schlenker
S
, Ulrich
HD
, Mayer
TU
and Jentsch
S.
A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly.
Cell.
1999;
96:
635
-644.
[PubMed]
.
-
9.
Hatakeyama
S
, Yada
M
, Matsumoto
M
, Ishida
N
and Nakayama
KI.
U box proteins as a new family of ubiquitin-protein ligases.
J Biol Chem.
2001;
276:
33111
-33120.
[PubMed]
.
-
10.
Kaneko
C
, Hatakeyama
S
, Matsumoto
M
, Yada
M
, Nakayama
K
and Nakayama
KI.
Characterization of the mouse gene for the U-box-type ubiquitin ligase UFD2a.
Biochem Biophys Res Commun.
2003;
300:
297
-304.
[PubMed]
.
-
11.
Cruciat
CM
, Hell
K
, Folsch
H
, Neupert
W
and Stuart
RA.
Bcs1p, an AAA-family member, is a chaperone for the assembly of the cytochrome bc(1) complex.
EMBO J.
1999;
18:
5226
-5233.
[PubMed]
.
-
12.
Golbik
R
, Lupas
AN
, Koretke
KK
, Baumeister
W
and Peters
J.
The Janus face of the archaeal Cdc48/p97 homologue VAT: protein folding versus unfolding.
Biol Chem.
1999;
380:
1049
-1062.
[PubMed]
.
-
13.
Kaneko-Oshikawa
C
, Nakagawa
T
, Yamada
M
, Yoshikawa
H
, Matsumoto
M
, Yada
M
, Hatakeyama
S
, Nakayama
K
and Nakayama
KI.
Mammalian E4 is required for cardiac development and maintenance of the nervous system.
Mol Cell Biol.
2005;
25:
10953
-10964.
[PubMed]
.
-
14.
Matsumoto
M
, Yada
M
, Hatakeyama
S
, Ishimoto
H
, Tanimura
T
, Tsuji
S
, Kakizuka
A
, Kitagawa
M
and Nakayama
KI.
Molecular clearance of ataxin-3 is regulated by a mammalian E4.
EMBO J.
2004;
23:
659
-669.
[PubMed]
.
-
15.
Okumura
F
, Hatakeyama
S
, Matsumoto
M
, Kamura
T
and Nakayama
KI.
Functional regulation of FEZ1 by the U-box-type ubiquitin ligase E4B contributes to neuritogenesis.
J Biol Chem.
2004;
279:
53533
-53543.
[PubMed]
.
-
16.
Bloom
L
and Horvitz
HR.
The Caenorhabditis elegans gene unc-76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation.
Proc Natl Acad Sci U S A.
1997;
94:
3414
-3419.
[PubMed]
.
-
17.
Conforti
L
, Tarlton
A
, Mack
TG
, Mi
W
, Buckmaster
EA
, Wagner
D
, Perry
VH
and Coleman
MP.
A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse.
Proc Natl Acad Sci U S A.
2000;
97:
11377
-11382.
[PubMed]
.
-
18.
Mack
TG
Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene.
Nat Neurosci.
2001;
4:
1199
-1206.
[PubMed]
.
-
19.
Borchelt
DR
, Davis
J
, Fischer
M
, Lee
MK
, Slunt
HH
, Ratovitsky
T
, Regard
J
, Copeland
NG
, Jenkins
NA
, Sisodia
SS
and Price
DL.
A vector for expressing foreign genes in the brains and hearts of transgenic mice.
Genet Anal.
1996;
13:
159
-163.
[PubMed]
.
-
20.
Hara
T
, Nakamura
K
, Matsui
M
, Yamamoto
A
, Nakahara
Y
, Suzuki-Migishima
R
, Yokoyama
M
, Mishima
K
, Saito
I
, Okano
H
and Mizushima
N.
Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice.
Nature.
2006;
441:
885
-889.
[PubMed]
.
-
21.
Komatsu
M
Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice.
Cell.
2007;
131:
1149
-1163.
[PubMed]
.
-
22.
Kim
MS
Hypothalamic localization of the feeding effect of agouti-related peptide and alpha-melanocyte-stimulating hormone.
Diabetes.
2000;
49:
177
-182.
[PubMed]
.
-
23.
Kublaoui
BM
, Holder
JL Jr
, Gemelli
T
and Zinn
AR.
Sim1 haploinsufficiency impairs melanocortin-mediated anorexia and activation of paraventricular nucleus neurons.
Mol Endocrinol.
2006;
20:
2483
-2492.
[PubMed]
.
-
24.
Sarkar
S
, Legradi
G
and Lechan
RM.
Intracerebroventricular administration of alpha-melanocyte stimulating hormone increases phosphorylation of CREB in TRH- and CRH-producing neurons of the hypothalamic paraventricular nucleus.
Brain Res.
2002;
945:
50
-59.
[PubMed]
.
-
25.
Wirth
MM
, Olszewski
PK
, Yu
C
, Levine
AS
and Giraudo
SQ.
Paraventricular hypothalamic alpha-melanocyte-stimulating hormone and MTII reduce feeding without causing aversive effects.
Peptides.
2001;
22:
129
-134.
[PubMed]
.
-
26.
Leibowitz
SF
, Hammer
NJ
and Chang
K.
Hypothalamic paraventricular nucleus lesions produce overeating and obesity in the rat.
Physiol Behav.
1981;
27:
1031
-1040.
[PubMed]
.
-
27.
Susaki
E
, Kaneko-Oshikawa
C
, Miyata
K
, Tabata
M
, Yamada
T
, Oike
Y
, Katagiri
H
and Nakayama
KI.
Increased E4 activity in mice leads to ubiquitin-containing aggregates and degeneration of hypothalamic neurons resulting in obesity.
J Biol Chem.
2010;
285:
15538
-15547.
[PubMed]
.
-
28.
Friedman
JM
and Halaas
JL.
Leptin and the regulation of body weight in mammals.
Nature.
1998;
395:
763
-770.
[PubMed]
.
-
29.
Elmquist
JK
, Elias
CF
and Saper
CB.
From lesions to leptin: hypothalamic control of food intake and body weight.
Neuron.
1999;
22:
221
-232.
[PubMed]
.
-
30.
Deng
HW
, Deng
H
, Liu
YJ
, Liu
YZ
, Xu
FH
, Shen
H
, Conway
T
, Li
JL
, Huang
QY
, Davies
KM
and Recker
RR.
A genomewide linkage scan for quantitative-trait loci for obesity phenotypes.
Am J Hum Genet.
2002;
70:
1138
-1151.
[PubMed]
.
-
31.
Stone
S
A major predisposition locus for severe obesity, at 4p15-p14.
Am J Hum Genet.
2002;
70:
1459
-1468.
[PubMed]
.