P66SHC and Ageing: ROS and TOR?
Abstract
Both Reactive Oxygen Species (ROS) and hyperactivation of the nutrient-sensing mTOR/S6 kinase cascade have been linked to aging and age-related diseases as well as to the anti-aging effect of calorie restriction. Recent findings that the pro-aging and pro-oxidant molecule p66shc contributes to S6K activation by nutrients and promotes insulin resistance and diabetes in mice may provide an answer to the "ROS or TOR?" dilemma.
In
late 90's, the idea that manipulation of one single gene could significantly
extend longevity of a complex model organism was certainly not an heretic one [1].
Successful attempts had already been made in worms and flies, not to speak
about budding yeast, whose unicellular simplicity makes it somehow close to a
cell culture system. It was already clear, in particular, that hypomorphic
mutations in the insulin/Igf1 (IIS) pathway could enhance lifespan in
Drosophila or C. Elegans, in a fashion that could mimic the effect of nutrient
restriction, for the longest time the most reliable model for laboratory
research on longevity. The role of
insulin/Igf signaling in mediating body response to nutrients, and the fact
that IIS is reduced by calorie restriction protocols, provided full rationale
to these observations, that brought to the notion that aging may represent,
rather than the uncontrolled catastrophe of the body, the product of a
genetically coded programme. Yet, report by Migliaccio and colleagues, that
mice lacking the 66kD isoform of the Shc (Src Homology and Collagen) protein
family lived 30% longer than p66-proficient littermates, caught the
scientific community by surprise, especially because p66KO mice, not only were
long lived, but appeared, unlike other murine models of longevity such as GH
deficient dwarf mice, phenotypically normal, fertile and healthy [2].
Shc proteins were known as adapter
molecules, i.e. signaling components deputed
to the assembly of macromolecular
complexes downstream of activated growth factor receptors (RTKs). A role for
SHCs in insulin signaling, in particular, had also been reported [3]. Thus, one
could have easily welcomed the p66KO mouse as the first (or one of the first)
mammalian example(s) of extended longevity by genetic attenuation of
insulin/Igf signaling. Another one, the Igf-1 receptor (IGF-1R) knock-out
mouse, was going to come shortly after [4].
Instead,
the linkage between p66 and longevity took an unexpected direction, becoming
one of the strongest arguments in support of the Harman's "free radical theory
of aging" [5]: in fact, p66- deficient mice and cells were found to present remarkably
reduced levels of ROS and increased resistance to oxidative stress.
Attenuation
of insulin signaling leads per se to reduced oxidative burden, by Daf-16/FoxO
dependent up-regulation of antioxidant defenses [6]; thus, the
oxidant-resistant phenotype of p66KO mice could still fit in the genetic model
of longevity centered on the insulin/Igf signaling cascade. Instead, second
surprise, solid biochemical studies revealed for p66shc a function completely
distinct from that of the other SHC proteins: it was found that, in response to
a number to pro-oxidant and apoptogenic stimuli, p66shc translocates to
mitochondria, where it directly generates reactive oxygen species, by
transferring electrons from cytochrome c to oxygen [7]. This finding tied p66
and its effect on longevity to ROS and mitochondria, in perfect agreement with
Harman's theories; accordingly, studies performed on p66KO mice involved p66 in
a number of typical age-related diseases, including vascular diabetic
complication and atherosclerosis, already suspected to be caused by excess
oxidative stress [8]. Interestingly, in keeping with initial predictions,
insulin signaling was indeed found to be defective in p66-deficient cells and
mice, but that was again related to the molecule's capacity to generate ROS,
that facilitate tyrosine kinase signaling by transient and reversible
inhibition of tyrosine phosphatases [9].
Accumulation
of cellular and tissue oxidative damage, however, may nor represent the only,
or even the most important, mechanism underlying body senescence and limitation
of lifespan. Mounting evidence indicate that effects of calorie restriction on
longevity involve a number of nutrient-sensing molecular networks that
regulate, beside ROS generation and scavenging, also DNA repair, inflammation,
cell proliferation and body growth (i.e. accumulation of biomass) [10]. One of
these evolutionarily conserved networks involves the sirtuin family of NAD+
dependent histone deacetylases (sirtuin 1 through 7 in mammals) that regulate
chromatin remodelling and gene transcription in response to cellular energy
status [11]. Another major nutrient-sensing pathway is centered on the TOR
(Target of Rapamycin) kinase and its downstream cascade. In mammalian cells,
m(ammalian)TOR regulates ribosomal protein synthesis, cell growth, cell cycle
progression, autophagy and mitochondrial function in response to the
availability of aminoacids and the intracellular levels of ATP. Additionally,
mTOR is activated by growth factor receptors. Including, of course, the insulin
receptor [12].
Several lines of evidence indicate that
nutrient and insulin-dependent regulation of TOR and its downstream cascade may
play a central role in aging and in the nutritional control of lifespan. In
yeast, flies and worms, hypomorphic mutations in this cascade extend longevity
[10]. Even more interestingly, the mTOR inhibitor Rapamycin extends lifespan
in mice and prevents age-related diseases [13], and so does genetic deletion of
the ribosomal S6 kinase (S6K), a major downstream effector of mTOR [14]. Thus,
inactivation of the mTOR pathway mimics the beneficial effect of calorie
restriction in rodents, clearly indicating that mTOR-dependent signaling
contributes to longevity determination by nutrients in mammals. Again, inhibition
of TOR may lead to increased antioxidant defenses, as observed in yeast and
flies [15], but could also promote autophagy and reduce intracellular
accumulation of pathologic proteins, that eventually leads to Endoplasmic
Reticulum (ER) stress and tissue aging [16]. Notably, accumulation of misfolded
proteins underlies typical senescence-associated pathologies like Alzheimer's
and vascular amyloidosis, while ER stress contributes to insulin resistance
and Metabolic Syndrome, another age-dependent disease [17].
Is
there a relationship between p66-dependent aging and the regulation of
longevity by nutrients, through the mTOR/S6K cascade? Or, in other words, does
the mTOR/S6K cascade contribute to p66 effects on mouse lifespan? Recent work
performed in our laboratory tried to address this seemingly relevant question
[18].
We
were initially interested in determining whether p66shc may have a role in
insulin resistance, the signaling dysfunction underlying glucose intolerance
and type 2 diabetes associated with overnutrition and overweight. The question
was legitimated by increasing evidence of a role for reactive oxygen species in
insulin desensitization [19], and by our previous observation of reduced liver
steatosis, a major inducer of insulin-resistance, in p66KO mice [20]. We indeed
found that obese (LepOb, leptin deficient) mice devoid of p66, although
gaining nearly as much weight as their p66-proficient littermates, remained
remarkably responsive to insulin and were significantly protected from
diabetes. Importantly, this finding correlated with reduced levels of
phosphorylation of S6K in the adipose tissue; additionally, isolated adipocytes
from p66KO obese mice displayed reduced S6K activity and preserved insulin
responsiveness compared to p66 WT cells, and p66KO preadipocytes were resistant
to the insulin-desensitizing effect of excess fatty acids in vitro.
These
findings fitted with the current model whereby excess nutrient (glucose and
Free Fatty Acids) and chronic hyperinsulinemia downregulate insulin response in
target tissues by hyperactivating S6K, that in turn leads to serine
phosphorylation and proteasomal degradation of the major insulin transducer
IRS-1 [21]. p66 would participate in this circuitry by somehow stimulating S6K.
Accordingly, we showed that overexpression of p66shc in 3T3L1 adipocytes leads
to hyperactivation of S6K and to hyperphosphorylation of IRS on serine
residues. Further molecular dissection of these biochemical events also
revealed that p66shc forms a complex with S6K 1 and IRS-1, thus facilitating
the signal-inibitory interaction between the two molecules. To our surprise,
these effects of p66 were largely independent from changes in the intracellular
redox state, or from the redox properties of p66 itself, but seemingly
explainable by the "traditional" function of p66shc as an adapter protein. We
concluded that p66shc, at least in adipocytes, promoted insulin and nutrient
signaling to S6K, and, consequently, the feed-back inhibitory action of S6K on
IRS-1, leading to diabetes in overfed animals.
While
these findings have obvious relevance for the understanding of signal
deregulation connecting obesity and overnutrition to diabetes, our observation
may add a novel perspective to the linkage between p66shc and lifespan
determination.
In fact, ablation of p66, by leading to reduced
responsiveness of S6K to nutrients, creates a Rapamycin-like (although
presumably milder) signaling block that conceivably promotes animal longevity, at least by preventing one major age-related disease, type 2
diabetes. In simpler words, p66 ablation could mimc calorie restricttion.
Notably type 2 diabetes recapitulates and accelerates many pathologic changes
(in vasculature, kidneys, eyes, peripheral nerves) that are typical of
senescence. These changes hit tissues that are largely insulin-independent for
their energy metabolism, but that are exposed to elevated amount of insulin and
glucose imposed by whole body insulin resistance. Interestingly, obese mice
lacking p66 live significantly longer than their p66WT controls (although less
than lean, WT mice) [17]. On the other hand, laboratory animals fed ad
libitum frequently develop overweight and glucose intolerance with age,
indicating that effects of p66shc observed in the context of genetic obesity
and diabetes may also be relevant to the aging process of non overtly obese
mice.
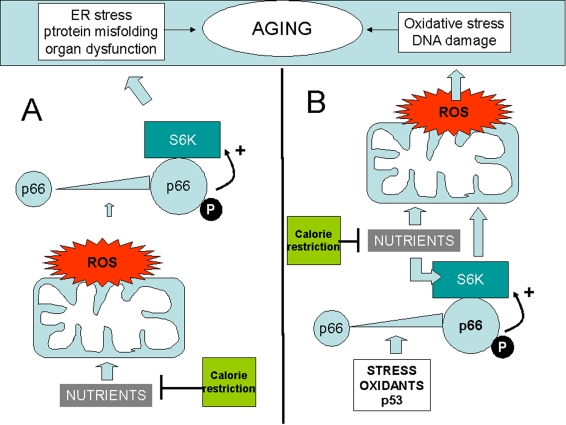
Figure 1. Two distinct models whereby p66shc may integrate ROS and the TOR/S6K cascade in the aging process.
(A) ROS upregulate p66shc and activate S6K through p66. Oxidant
species could be generated by mitochondria in response to nutirents, thus
creating an alternative route for nutrient sensing by S6K. (B) S6K,
activated by p66, increases ROS formation in mitochondria. In this case p66
could be in turn activated by cellular stress, by p53, or by
environmental oxidants. In both examples p66 effects on aging are
inhibited by calorie restriction (green box) that reduces nutrient supply.
Activation" of p66shc is depicted as a result of increased expression
(larger icon) and serine phophorylation (letter "P"). Both changes have in
fact been reported in response to diverse stresses in mammalian cells.
Apart
from prevention of glucose dysmetabolism, all the S6K-related mechanisms for
lifespan extension may operate, in view of our findings, in p66KO mice. For
instance, reduced protein translation may attenuate ER stress in critical
tissues and reduce progression and severity of age-related diseases due to
accumulation of misfolded proteins. While this possibility deserves to be
tested in appropriate model systems (such as mice prone to Alzheimer's disease
crossed to p66KO mice), we have preliminary evidence that overexpression of
p66shc in preadipocites and kidney cells increases ER stress in parallel with
hyperactivation of S6K.
Along
similar lines, increased autophagy, due to S6K attenuation, may contribute to
the long-lived phenotype of p66 deficient animals, another possibility to be
verified.
Finally,
prevention of cancer contributes to lifespan extension by calorie restriction
and S6K blockade. This may be true also in p66KO mice. Interestingly, in spite
of p66shc operating in the p53-initiated apoptotic pathway [22], no increase in
tumor incidence has been described in this mouse strain. Based on our
prediction such incidence may even be lower than in wild type animals, due, at
least in part, to reduced mTOR/S6K signaling in cancer cells. This is again a
testable hypothesis.
Can
these views be reconciled with current, "ROS-centric" model for lifespan
limitation by p66 [23]? In
principle, ROS can operate both upstream and downstream of the TOR cascade.
In one scenario, p66 action on S6K may lead to increased mitochondrial
metabolism and as a consequence to a rise of mitochondrial ROS [24], as
observed in cells were p66shc is overexpressed [2]. In simple terms, mTOR/S6K
may mediate, at least in part, the pro-oxidant action of p66 (Figure 1B).
More intriguingly, ROS may act upstream
of the p66/S6K module, since p66shc not only generates ROS, but is also
stimulated by oxidants [2]. For instance, in fibroblasts exposed to oxidative
stress, PI3K/AkT activation by ROS is mediated, at least to some extent, by
p66shc [25]; AkT can, in turn, activate mTOR. ROS are also generated in
mitochondria in response to energy substrates; these species may increase the
phospho-rylation/expression level of p66, thereby promoting its
(redox-independent) stimulatory action on S6K. This would represent an
intriguing alterantive route for nutrients to signal, via mitochondria,
ROS and p66shc, to the mTOR/S6K cascade (Figure 1A). Of note, phosphorylation
of p66, a modification that correlates with its biological activity, was found
to be increased in pre-adipocytes exposed to hyperglycemia or excess FFA, as if
p66 were actually behaving as a sensor of nutrient abundance in these
cellular contexts [17].
In
all the above scenarios, p66, S6K and ROS lie on the same nutrient sensitive
pathway, mechanistically linked to aging and potentially targetable by calorie
restriction (Figure 1 A and B).
In
conclusion, the observation that p66shc contributes to S6K activation in
response to glucose, amino acids and insulin, supports the concept that aging
and age-related diseases are driven by TOR (not by ROS) and p66sch accelerates
aging by activating TOR [26]; revealing the existence of a novel
nutrient-regulated pathway to senescence, in which p66shc works as an adaptor
(what else?) between ROS and TOR.
Acknowledgments
The author of this manuscript has no conflict of
interests to declare.
Conflicts of Interest
The author of this manuscript has no conflict of
interests to declare.
References
-
1.
Guarente
L
and Kenyon
C.
Genetic pathways that regulate aging in model organisms.
Nature.
2000;
408:
255
-262.
[PubMed]
.
-
2.
Migliaccio
E
, Giorgio
M
, Mele
S
, Pelicci
G
, Reboldi
P
, Pandolfi
PP
, Lanfrancone
L
and Pelicci
PG.
The p66shc adaptor protein controls oxidative stress response and life span in mammals.
Nature.
1999;
402:
309
-313.
[PubMed]
.
-
3.
Giorgetti
S
, Pelicci
PG
, Pelicci
G
and Van
Obberghen E.
Involvement of Src homology/collagen (SHC) proteins in signaling through the insulin receptor and the insulin-like-growth-factor-I-receptor.
Eur J Biochem.
1994;
223:
195
-202.
[PubMed]
.
-
4.
Lithgow
GJ
and Gill
MS.
Physiology: Cost-free longevity in mice.
Nature.
2003;
421:
125
-126.
[PubMed]
.
-
5.
Harman
D
A biologic clock: the mitochondria.
Journal of the American Geriatrics Society.
1972;
20:
145
-147.
[PubMed]
.
-
6.
Kops
GJ
, Dansen
TB
, Polderman
PE
, Saarloos
I
, Wirtz
KW
, Coffer
PJ
, Huang
TT
, Bos
JL
, Medema
RH
and Burgering
BM.
Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress.
Nature.
2002;
419:
316
-321.
[PubMed]
.
-
7.
Giorgio
M
, Migliaccio
E
, Orsini
F
, Paolucci
D
and Moroni
M.
. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis.
Cell.
2005;
122:
221
-233.
[PubMed]
.
-
8.
Cosentino
F
, Francia
P
, Camici
GG
, Pelicci
PG
, Lüscher
TF
and Volpe
M.
Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein.
Arterioscler Thromb Vasc Biol.
2008;
28:
622
-628.
[PubMed]
.
-
9.
Berniakovich
I
, Trinei
M
, Stendardo
M
, Migliaccio
E
and Minucci
S.
p66Shc-generated oxidative signal promotes fat accumulation.
J Biol Chem.
2008;
283:
34283
-34293.
[PubMed]
.
-
10.
Fontana
L
, Partridge
L
and Longo
VD.
Extending healthy life span--from yeast to humans.
Science.
2010;
328:
321
-326.
[PubMed]
.
-
11.
Guarente
L
Sirtuins in aging and disease.
Cold Spring Harb Symp Quant Biol.
2007;
72:
483
-438.
[PubMed]
.
-
12.
Sarbassov
DD
, Ali
SM
and Sabatini
DM.
Growing roles for the mTOR pathway.
Curr Opin Cell Biol.
2005;
17:
596
-603.
[PubMed]
.
-
13.
Harrison
DE
, Strong
R
, Sharp
ZD
, Nelson
JF
and Astle
CM.
Rapamycin fed late in life extends lifespan in genetically heterogeneous mice.
Nature.
2009;
406:
392
-396.
[PubMed]
.
-
14.
Selman
C
, Tullet
JM
, Wieser
D
, Irvine
E
, Lingard
SJ
, Choudhury
AI
, Claret
M
, Al-Qassab
H
, Carmignac
D
, Ramadani
F
, Woods
A
, Robinson
IC
, Schuster
E
, Batterham
RL
, Kozma
SC
, Thomas
G
, Carling
D
, Okkenhaug
K
, Thornton
JM
, Partridge
L
, Gems
D
and Withers
DJ.
Ribosomal protein S6 kinase 1 signaling regulates mammalian life span.
Science.
2009;
326:
140
-144.
[PubMed]
.
-
15.
Bjedov
I
, Toivonen
JM
, Kerr
F
, Slack
C
, Jacobson
J
, Foley
A
and Partridge
L.
Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster.
Cell Metab.
2010;
11:
35
-46.
[PubMed]
.
-
16.
Gregersen
N
, Bross
P
, Vang
S
and Christensen
JH.
Protein misfolding and human disease.
Annu Rev Genomics Hum Genet.
2006;
7:
103
-124.
[PubMed]
.
-
17.
Ozcan
U
, Cao
Q
, Yilmaz
E
, Lee
AH
, Iwakoshi
NN
, Ozdelen
E
, Tuncman
G
, Görgün
C
, Glimcher
LH
and Hotamisligil
GS.
Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes.
Science.
2004;
306:
457
-461.
[PubMed]
.
-
18.
Ranieri
SC
, Fusco
S
, Panieri
E
, Labate
V
, Mele
M
, Tesori
V
, Ferrara
AM
, Maulucci
G
, De
Spirito M
, Martorana
GE
, Galeotti
T
and Pani
G.
Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance.
Proc Natl Acad Sci U S A.
2010;
107:
13420
-13425.
[PubMed]
.
-
19.
Houstis
N
, Rosen
ED
and Lander
ES.
Reactive oxygen species have a causal role in multiple forms of insulin resistance.
Nature.
2006;
440:
944
-948.
[PubMed]
.
-
20.
Koch
OR
, Fusco
S
, Ranieri
SC
, Maulucci
G
, Palozza
P
, Larocca
LM
, Cravero
AA
, Farre'
SM
, De
Spirito M
, Galeotti
T
and Pani
G.
Role of the life span determinant P66(shcA) in ethanol-induced liver damage.
Lab Invest.
2008;
88:
750
-760.
[PubMed]
.
-
21.
Um
SH
, D'Alessio
D
and Thomas
G.
Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1.
Cell Metab.
2006;
3:
393
-402.
[PubMed]
.
-
22.
Trinei
M
, Giorgio
M
, Cicalese
A
, Barozzi
S
and Ventura
A.
A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis.
Oncogene.
2002;
21:
3872
-3878.
[PubMed]
.
-
23.
Trinei
M
, Berniakovich
I
, Beltrami
E
, Migliaccio
E
, Fassina
A
, Pelicci
P
and Giorgio
M.
P66Shc signals to age.
Aging.
2009;
1:
503
-510.
[PubMed]
.
-
24.
Schieke
SM
and Finkel
T.
Mitochondrial signaling, TOR, and life span.
Biol Chem.
2006;
387:
1357
-1361.
[PubMed]
.
-
25.
Nemoto
S
and Finkel
T.
Redox regulation of forkhead proteins through a p66shc-dependent signalling pathway.
Science.
2002;
295:
2450
-2452.
[PubMed]
.
-
26.
Blagosklonny
MV
Aging: ROS or TOR.
Cell Cycle.
2008;
7:
3344
-3354.
[PubMed]
.