Abstract
The combination of functional genomics with next generation sequencing facilitates new experimental strategies for addressing complex biological phenomena. Here, we report the identification of a gain-of-function allele of peroxiredoxin (thioredoxin peroxidase, Tsa1p) via whole-genome re-sequencing of a dominantSaccharomyces cerevisiae mutant obtained by chemical mutagenesis. Yeast strain K6001, a screening system for lifespan phenotypes, was treated with ethyl methanesulfonate (EMS). We isolated an oxidative stress-resistant mutant (B7) which transmitted this phenotype in a background-independent, monogenic and dominant way. By massive parallel pyrosequencing, we generated an 38.8 fold whole-genome coverage of the strains, which differed in 12,482 positions from the reference (S288c) genome. Via a subtraction strategy, we could narrow this number to 13 total and 4 missense nucleotide variations that were specific for the mutant. Via expression in wild type backgrounds, we show that one of these mutations, exchanging a residue in the peroxiredoxin Tsa1p, was responsible for the mutant phenotype causing background-independent dominant oxidative stress-resistance. These effects were not provoked by altered Tsa1p levels, nor could they be simulated by deletion, haploinsufficiency or over-expression of the wild-type allele. Furthermore, via both a mother-enrichment technique and a micromanipulation assay, we found a robust premature aging phenotype of this oxidant-resistant strain. Thus, TSA1-B7 encodes for a novel dominant form of peroxiredoxin, and establishes a new connection between oxidative stress and aging. In addition, this study shows that the re-sequencing of entire genomes is becoming a promising alternative for the identification of functional alleles in approaches of classic molecular genetics.
Introduction
The free radical
theory of aging implies that oxidative stress, and the generation of free
radicals, are causally involved in the process of aging [1-3].
This theory is supported
by many observations, including that yeast mother cells retain oxidatively
damaged macro-molecules, whereas the daughter cells are formed from a juvenile
set of proteins [4], or inherit
functional enzymes whereas the damaged species are retained with the mother [5]. However,
other discoveries challenge a causal implication of free radicals in different
aging phenotypes [6]. Although
most long-living mutants of S. cerevisiae and C. elegans tolerate
high doses of oxidants, not all oxidant-resistant mutants are long living. As
an example, both deletion of the metabolic regulator AFO1 [7] and reduced
activity of the metabolic enzyme TPI1 [8] increase
oxidant tolerances of yeast, but Δafo1 cells are massively long-living whereas TPI-deficient
cells have a strong premature aging phenotype [7, 8]. In
drosophila, mitochondrial ROS production correlates with aging but conversely,
is not sufficient to alter lifespan [9]. All these
observations are further complicated by the fact that mutants which are
long-living under one environment/condition do not necessarily show this
phenotype under other circumstances, i.e. yeast mutants with prolonged survival
at 4°C are not enriched for mutants that are long-living at a higher
temperatures [10]. In
addition, there is a close relation of growth rate, metabolic activity and
aging [11]. Since
metabolism is itself a primary source for free radicals within the cell, it is
difficult to distinguish between consequence and causality of oxidative damage
during the aging process [6, 11]. Thus,
although oxidative stress and free radicals are important players, their exact
role during aging and the complex interplay of the involved genetic and
biochemical components has yet to be clarified.
Systematic functional genetics/genomics is powerful in
the identification of genetic components of biological processes and their
interactions. With the introduction of systematic genetic libraries, such as an
entire knock-out library of the yeast S. cerevisiae one decade ago [12], a new era of functional
genetics began. Screening of systematic libraries allows circumventing the most
time-consuming and limiting step of experimental genetics, which is the
identification of the functional mutation. Screens with the systematic
libraries identified many components that influence yeast aging phenotypes [10, 13, 14]. However, all pre-made libraries
have the disadvantage of being limited in the number of alleles they contain.
In practical terms, genome-wide knock-out, over-expression and copy number
variation libraries can be generated, but nothing such as genome-wide
‘point-mutation' libraries which would allow the isolation of alleles with new
functionality. Therefore, although highly efficient, screens with systematic
genetic libraries miss functional connections that can be identified by the isolation
of new alleles in random-mutagenesis screenings. Thus, there is demand for
experimental strategies that increase the efficiency of these classic
approaches.
Here, we provide a case where
whole-genome re-sequencing led to the identification of a new and dominant
peroxiredoxin allele that causes oxidative stress resistance and premature
aging. The W303-derived yeast strain K6001 [15], a model
system that allows the enrichment of yeast mother cells [16], was used
in a random mutagenesis experiment to isolate a mutant that exhibited strong
resistance to the chemical oxidant cumene hydroperoxide (CHP). The mutant was
dominant and segregated this phenotype in a regular way and independent of
genetic background, observations which pointed to a monogenic trait. Using
whole-genome re-sequencing of both strains by the Roche/454 system, and
comparison of their genomes, we identified the mutation as a single nucleotide
exchange in the gene, TSA1, coding for thioredoxin peroxidase
(peroxiredoxin).
Results
Random
mutagenesis and isolation of an oxidant-resistant K6001 mutant
Cell
division of S. cerevisiae is asymmetric; mother and daughter cells are
clearly recognizable by their size. Mother cells can complete a limited number
of cell cycles. At the end of this so-called replicative lifespan, the
cells persist for some further time in a non-dividing state, and finally die,
predominantly by apoptosis [17]. The yeast
strain K6001 has been used to study this phenotype, because it facilitates the
selective enrichment of yeast mother cells. On glucose media, daughters of
K6001 do not express the essential CDC6 gene, and are therefore unable
to complete their cell cycle. Thus, the biomass which a K6001 culture achieves
in glucose media, is a function of the number of daughters per mother and
indicates its average replicative lifespan [16].
To
identify new genetic connections between aging and oxidative stress, we used
K6001 in a random mutagenesis experiment. An exponential K6001 culture was
mutagenized with ethyl methansulfonate (EMS) and grown for two generations
under non-selective conditions to enable mutation fixation. Mutagenized cells
were then plated onto agar that contained the synthetic oxidant cumene
hydroperoxide (CHP) at a concentration lethal for the wild-type strain. For
every million of cells plated, an average of three colonies gained resistance
and grew on the CHP-containing media. Obtained stress resistant mutants were
tested for their replicative lifespan. We are presenting here one mutant
(K6001-B7) that displayed resistance/sensitivity to multiple oxidants and clear
regular 2:2 segregation in genetic crosses. The isogenic mating partner, K6001α, was obtained by mating type switching of K6001 induced by a plasmid
carrying the wild type HO gene. The mutant was mated to K6001α and the diploids were sporulated. Tetrads gained by sporulation were
tested for resistance to CHP, t-butyl hydroperoxide (t-BHT), diamide, and
hydrogen peroxide. Figure 1 summarizes the oxidant phenotypes of K6001-B7, the
diploid obtained in the outcross just described, and the haploid progeny
derived thereof (one typical tetrad). The mutant strain K6001-B7, the diploids
obtained by outcrossing it to
wild type, and the haploid progeny all showed the same phenotype: strong
resistance to CHP, increased resistance to t-BHT, sensitivity to hydrogen
peroxide (Figure 1A), and normal growth in the presence of diamide, the trait
was inherited in a monogenic way (2:2 segregation in tetrads; Figure 1B) and
the mutant allele was dominant even in crosses with a non-related wild type
strain, BY4742 (Figure 1C lower panel).
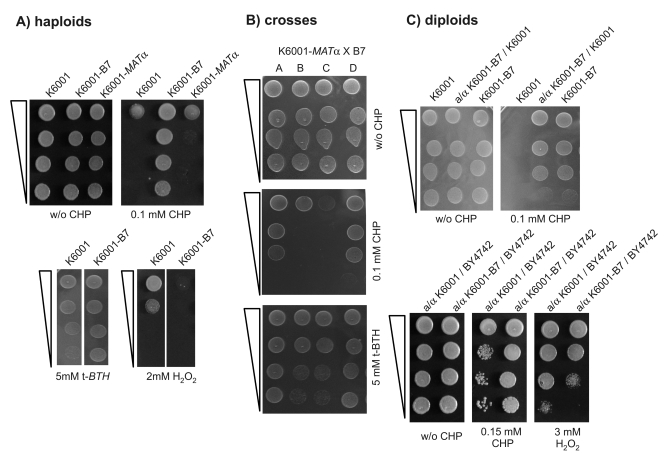
Figure 1. K6001-B7 carries a monogenic and dominant allele that confers resistance to cumene hydroperoxide (CHP). (A) K6001-B7 is
resistant to CHP. Overnight cultures of the parent (K6001, MATa),
K6001-B7 and an isogenic MATα
(K6001-α32) strain were
spotted as serial dilution on SC agar with and without CHP and grown at
28°C (upper panel). K6001-B7 grows on 0.1 mM CHP, a concentration lethal
for K6001 and K6001-α32. (lower
panel) Similar test were performed with oxidants tert-butyl hydroperoxide
(t-BTH) (left) and H2O2 (right). (B) K6001-B7
transmits with a monogenic Mendelian trait. K6001-B7 was mated with
K6001-α32, tetrads
gained by sporulation and tested for CHP and t-BTH resistance. Shown is the
2:2 segregation of a representative tetrade. (C) B7 is a dominant
mutation. (upper panel) K6001 and K6001-B7 were mated with
K6001-α32, resultant
diploids assayed for CHP resistance. +/B7 diploids retained the stress
resistance of the B7 haploid. (lower panel) B7 is dominant across
backgrounds. K6001 and K6001-B7 were mated with the distant S.
cerevisiae strain BY4742. Similar to the pure K6001 background
situation, BY/K6001-B7 diploids were more resistant to CHP (left) and
sensitive to H2O2 (right) compared to the BY/K6001
diploids.
Identification
of the K6001-B7 gene via whole-genome re-sequencing
Dominant
mutations yielding stress-resistance are difficult to identify using classic
yeast genetics; e.g. resistance phenotypes are commonly observed when
anti-oxidant factors are over-expressed or over-active, leading to dominance of
the mutant allele and limiting the possibilities and the specificity of
complementation strategies with clone libraries that often over-express the
inserted genes, even if they are centromeric. Furthermore, for yeast strains
evolutionary distant from the reference genome of S288c, such as W303, the use
of whole-genome-tiling arrays is limited due to potential unspecific
hybridisation with primers designed for this distant genome.
Here, we decided on a whole-genome
resequencing strategy using the Roche/454 platform to identify the B7 mutation.
For this, we isolated genomic DNA from both K6001 and B7 and generated 454
sequencing libraries. Libraries were quality- controlled and sequenced by a
Titanium sequencing kit (Roche). The run produced 662 MB of high-quality
sequence at a median read-length of 527 bp (average read-length 499 bp). First,
we aligned the sequence information to the S288c reference genome. 96.89 % of
the coding and 96.83% of the noncoding regions were called at high quality in
K6001 and 96,97%/96,98% in K6001-B7 (merged 97,36%/97,54%). Most of the
non-called regions were neither sequenced in K6001 nor in K6001-B7, indicating
that they were physically absent. (see Supplementary Figure 1 for an
illustration of coverage uniformity). Based on this data, we calculated an
average 19.2 fold coverage for K6001 and 19.5 fold coverage for K6001-B7. To
compare the K6001/K6001-B7 genome with the S288c reference, we merged both
sequence runs for an alignment, yielding an 38.8 fold total coverage. At this
depth, we detected 12,482 SNVs and small (<50bp) insertions and deletions
(that were sequenced at a minimum of 3x and on both strands) which
distinguished K6001 from the reference genome. Surprisingly, although
sequencing a haploid genome, we detected several mutations that had a calling
rate between 5% and 95%. In a diploid genome, one would assume that these
variants are heterozygotes. However, since we do not expect the existence of
true heterozygotes in a haploid genome, we refer to these variants as presently
unexplained non-uniform sequences. These variants were not entirely randomly
distributed in the genome, many of these (62%) clustered close to the telomeric
regions. This result could point to a natural variability of the genome in
these regions. However, the result that the B7 phenotype was segregating in 2:2
manner allowed the exclusion
of these non-uniform SNVs from the candidate list. By setting a threshold of a
minimal calling rate >95%, we reduced the list to 7213 uniform SNVs and
small insertion/deletions.
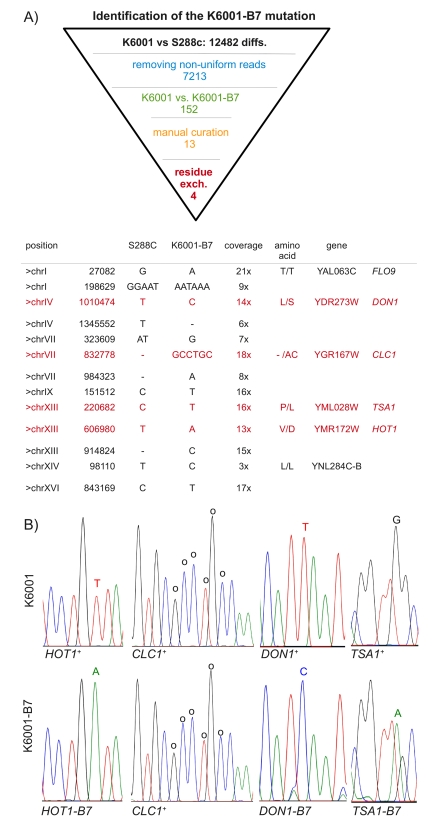
Figure 2. Identification of K6001-B7 by subtractive whole-genome resequencing. (A)
454 sequencing found 12,484 genetic differences between K6001 and the
S288c reference genome. Four K6001-B7 candidates were identified by
systematic narrowing of this list via the exclusion of non-uniform reads,
substracting K6001 from K6001-B7, and the manual exclusion of alignment
artefacts. Four of the remaining 13 variants were predicted to result in
amino acid exchange. (B) Sanger resequencing of candidate regions.
Mutant variants are highlighted. Please note that for TSA1 the
sequence trace of the reverse strand is shown.
We
continued our investigations by comparing K6001 with B7 by removing all
variants which were found in both genomes. This subtractive analysis narrowed
our candidate list to 152 K6001-B7 specific mutations. Finally, we were able to
remove alignment artefacts of the mapping algorithm by manual curation;
primarily, some two-nucleotide transversions were wrongly called. In addition,
we excluded all nucleotide variations which distinguished K6001-B7 from the
reference genome, but were simply not sequenced at sufficient quality in K6001
and therefore potential false-positives.
The
final list contained 13 candidate mutations. As illustrated in Figure 2, four
of these were predicted to cause amino acid exchanges. We amplified these regions by PCR and subjected them to Sanger sequencing.
The three SNV resulting in residue exchanges could be verified by
capillary re-sequencing (Figure 2B). The fourth difference, a six nucleotide
insertion of the CLC1 gene, was also real, but found in both the K6001
and K6001-B7 strain.
Oxidant
resistance of B7 is mediated by a single residue exchange within the TSA1 gene
To
identify the B7 gene among these candidates, we chose a classic strategy of cloning and phenotypic analysis.
The four potential candidate genes were PCR-amplified from K6001-B7 genomic
DNA, and sub-cloned into a yeast single-copy (centromeric) expression vector
along with their endogenous promoter sequences. The plasmids subsequently
verified by sequencing were transformed into K6001, and monoclonal descendants
selected on SC Galmedia lacking histidine. As illustrated in Figure 3A, the transformants ectopically expressing HOT1-B7, CLC1-B7, DON1-B7
as well those harbouring the control plasmids were viable on SC-His/Galmedia,
but not more resistant to CHP than the wild type.
Table 1. Oligonucleotide primers.
| CDS | fwd oligo | rev oligo | cloning |
| DON1 |
TAGAATTC
AGGGTACAGGCGAAGAAATG
|
TAGTCGAC
CTACGTAAAACTTAATTCTT
| EcoRI/SalI
|
| CLC1 |
GAAGAGCT
CAACAATACAATAAACCCAATC
|
TGGTCGAC
TTAAGCACCGGGAGCCTTCG
| SacI/SalI
|
| TSA1 |
GAGAGCTC
ATACGCTACCCAAGTACAGAAG
|
TGTCTCGAG
TTATTTGTTGGCAGCTTCGA
| SacI/XhoI
|
| Hot1 |
GAGAGCTC
ATTATATCCATGTTAAGTTCG
|
TATCTCGAG
CTATATTCCAGCAAGGCTCT
| SacI/XhoI
|
|
Underlined
sequences represent introduced restriction sites
| |
Table 2. MRM transitions.
| Q1/Q3
transition
| sequence
| Charge/Fragment
ion
|
| TSA-pep_1
| 617.85
- 984.55
| HITINDLPVGR
| 2+
/ y9
|
| TSA-pep_2
| 451.77
- 732.43
| GLFIIDPK
| 2+
/ y6
|
| TPI-pep_1
| 762.37
-989.49
| ASGAFTGENSVDQIK
| 2+
/ y9
|
| TPI-pep_2
| 758.93
- 864.46
| KPQVTVGAQNAYLK
| 2+
/ y8
|
In
contrast, the transformants of the TSA1-B7 encoding plasmid grew
perfectly well on media containing 0.05 mM CHP. To exclude in a second step,
that this phenotype was the result of a gene dose effect caused by the extra
copy of TSA1, we generated an additional, isogenic plasmid encoding for
its wild type form as well. Along with the empty as well as the TSA1-B7
vector, this plasmid was transformed into K6001 and the S288c derived BY4741
strain. Selected trans-formants were grown over night, and spotted as dilution
series on agar plates with and without the oxidant. As illustrated in Figure 3B, only yeast containing the TSA1-B7 plasmid, but not its wild-type
form, were resistant to CHP. This phenotype was also observed in the S288c
(BY4741) background, confirming the dominant and background-independent
inheritance. Thus, a new dominant allele in B7, encoding for Tsa1pPro182Leu,
was responsible for the increased oxidant tolerance of the K6001-B7 mutant.
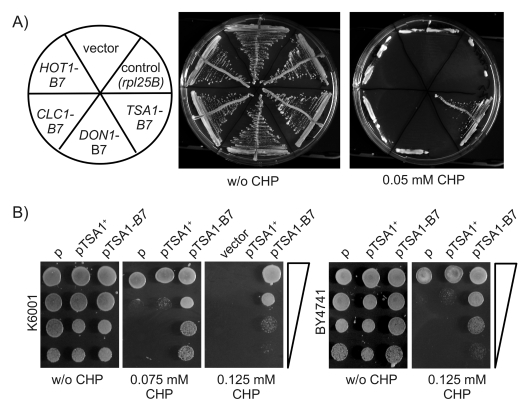
Figure 3. Ectopic expression of TSA1-B7 mediates CHP resistance. (A) Candidate
genes HOT1, CLC1, DON1, and TSA1 were amplified from the
K6001-B7 genome, subcloned, and ectopically expressed in the K6001 parent.
Transformants expressing TSA1-B7 were viable on CHP media. (B)
CHP resistance is specific to the expression of TSA1-B7. Centromeric
plasmids encoding wild type TSA1+ and its B7 form were
transformed into K6001. The additional copy of TSA1+ had
no effect on CHP resistance in K6001 (left) and S288c/BY4741 (right).
To
gain insights, if TSA-B7 represents a gain or a loss of function allele,
we first assayed TSA1-B7 mRNA levels in the K6001 parent and its B7
mutant. As shown in Figure 4A, via quantitative RT-PCR, we could not detect any
difference in TSA1 mRNA expression between the K6001 parent and
K6001-B7. Next, using targeted mass spectrometry, we addressed Tsa1p protein
levels. By quantifying two Tsa1p-specific tryptic peptides (GLFIIDPK and
HITINDLPVGR) and nor-malization of their peak areas to peptides specific for
Triosephosphate isomerase (ASGAFTGENSVDQIK and KPQVTVGAQNAYLK), we found that
Tsa1p is expressed at identical levels in K6001 and K6001-B7 (Figure 4B).
Thus, stress resistance caused by TSA1-B7 is not attributable to altered
expression of Tsa1p.
Next, we questioned, if the stress
resistance of B7 might be attributable to loss of function of Tsa1p. As shown
in Figure 4C left, deletion of the TSA1 gene in two haploid yeast
strains decreased rather then increased yeast's resistance to CHP. Thus,
depletion of TSA1 does not result in
the TSA-B7 phenotype, indicating that the dominantTSA1-B7 is not a loss of function allele. Next, we spotted diploid
wild-type, TSA1/Δtsa1 heterozygous and Δtsa1/Δtsa1 homozygous strains on oxidant-containing agar.
Whereas the homozygous Δtsa1 / Δtsa1 deletion strain was sensitive to CHP, this phenotype
was not detected in the TSA1/Δtsa1 heterozygotes (Figure 4C, right). Thus, partial loss
of TSA1 due to a haploinsufficiency does not resemble the B7
phenotype.
We
continued by addressing the effects of ectopic Tsa1 and Tsa1-B7 expression in
the Δtsa1
background. BY4741 based Δtsa1 yeast was transformed with TSA1- and TSA1-B7-
as well as empty single-copy (CEN) plasmids, and assayed for CHP resistance.
Expression of TSA1 in the deficient background restored the wild-type
phenotype, whereas B7 expression resulted in CHP resistant transformants (Figure 4D).
Finally, we questioned if the increased CHP resistance
might be attributable to higher Tsa1 activity and thus, could be simulated by
overexpression of wild-type Tsa1p. We subcloned TSA1 and TSA-B7
into a high-copy 2μ plasmid (p423GPD) and transformed both K6001 and K6001-B7.
As shown in Figure 4E, overexpression of wild-type TSA1 did not simulate
the effects of the B7 mutation. Unexpectedly, however, wild-type K6001 cells
harbouring the multicopy overexpression plasmid for TSA1-B7 resulted in
less CHP resistant cells than single-copy counterparts (Figure 4E left). This
effect was not seen in the mutant background (Figure 4E right). However, here
similar effects were caused by multicopy expression of the wild-type allele.
Thus, heterogeneous overexpression of TSA1 with TSA1-B7 reduces
the stress resistance mediated by the TSA1-B7 allele.
In sum, these results indicate that TSA1-B7 is a gain
of function allele. TSA1-B7 is dominant above TSA1 across yeast
backgrounds, its phenotype is not simulated by TSA1 over- or
underexpression nor its deletion, and finally, heterogeneous TSA1/TSA1-B7
overepxression diminishes the stress resistant phenotype.
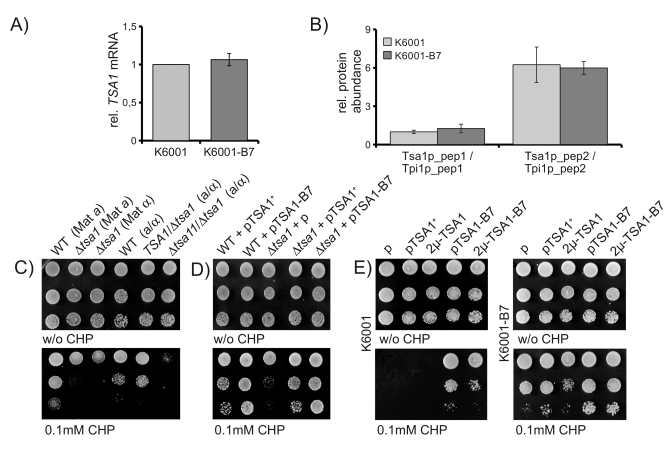
Figure 4. CHP resistance is mediated by a TSA1 gain of function. (A) TSA1
mRNA expression does not differ between K6001 and K6001-B7. TSA1 mRNA
levels were assayed by classic qRT-PCR and normalized to the geomean of
reference transcripts ACN9, ATG27 and TAF10. (B) Tsa1p
protein levels do not differ between K6001 and K6001-B7. Two Tsa1p
specific tryptic peptides were quantified by nanoLC-MS/MS and set in
relation to two peptides of triosephosphate isomerase (Tpi1p) in both K6001
and K6001-B7 extracts. (C) Deletion of tsa1, but not TSA1/Δtsa1heterozygosity,
decreases CHP tolerance. Tsa1 deletion strains of MATa and MATα mating types, as well as
corresponding heterozygote and homozygote diploids of the S288c background,
were spotted on CHP containing agar and grown at 28°C. (D) CHP
sensitivity of Δtsa1 is restored
upon ectopic expression of TSA1+. MATa Δtsa1 cells were
transformed with single-copy expression vectors encoding TSA1+ and TSA1-B7
and assayed for growth on CHP containing media. (E) Mulicopy
overexpression of TSA1+ does not mimic TSA1-B7, heterogeneous
overexpression of TSA1+/B7diminishes the oxidant resistance
phenotype. K6001 (left) and K6001-B7 (right) were transformed with
single-copy (p) or high-copy (2μ) TSA1+ and TSA1-B7
expression plasmids, spotted on CHP containing yeast agar, and grown at
28°C for three days.
K6001-B7 has a premature aging phenotype
Since altered resistance to oxidants has often been
observed in yeast strains with aging phenotypes, we screened for potential
alterations in the replicative lifespan of the B7 mutant. K6001 offered the
possibility to screen for alterations replicative aging via the generation of
growth curves in galactose- and glucose, an alternative to time-consuming
micromanipulation experiments [16]. As illustrated in Figure 5A,
K6001 and K6001-B7 both showed a comparable doubling time in galactose.
However, when shifted to glucose, linear growth stagnated at a lower biomass in
K6001-B7. The final biomass reached by the wild type and mutant was
significantly different. This indicated that - although oxidant-resistant -
K6001-B7 has a premature aging phenotype. To confirm this result, we assayed
replicative aging of K6001 via removal and counting of daughter cells by
micromanipulation (Figure 5B). On galactose media, the median lifespan of K6001 was 17 generations, and
the lifespan of K6001-B7 shortened to 13 generations.
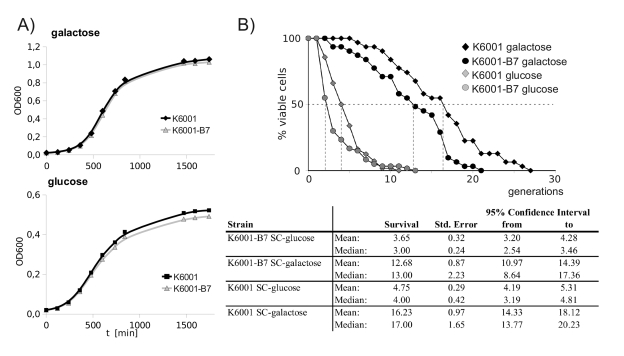
Figure 5. K6001-B7 has a shortened replicative lifespan. (A) K6001 and
K6001-B7 have similar growth in galactose (upper panel), but K6001
stagnates at a lower biomass in glucose media (lower panel). (B) Lifespan
assay by micromanipulation. Daughter cells from K6001 and K6001-B7 were
continuously removed by micormanipulation and counted (upper panel) and
analyzed statistically (lower panel). The shortened lifespan of K6001-B7 in
both glucose and galactose media was tested statistically significant using
Mantel-Cox, Breslow as well as Tarone/Ware statistics.
The
genetic manipulations of K6001 dramatically shorten its lifespan on glucose
media [16].
Nonetheless, we also compared the lifespan of K6001 and K6001-B7 mothers under
these conditions. Here, the median replicative lifespan was 3 for K6001-B7, and
4 generations for K6001. Thus, the premature aging phenotype of K6001-B7 is not
galactose-specific. Differences in the aging phenotypes on both media were
statistically significant as tested by Log Rank-, Breslow- and Tarone Ware
statistics.
Discussion
Here,
we describe the generation, identification and initial functional analysis of a
dominant peroxiredoxin allele which causes oxidative stress resistance and
premature aging in yeast. The oxidant-resistant mutant was isolated after EMS
treatment of the yeast aging model strain K6001, a descendant of the broadly used
yeast strain W303. Subsequent experiments demonstrat-ed that B7 is transmitted
in a monogenic and dominant pattern.
The
classical genetic and molecular genetic approach for the dissection of a
dominant monogenic trait is constructing a genomic clone bank representative
for the genome of the mutant, transforming this bank into wild type cells,
selecting for clones that show the phenotype (in our case, CHP resistance),
cloning and subcloning, and identifying the gene by capillary sequencing (see [18] for an
overview on classic screenings methods). For the case presented here, this
strategy would be burdened by several limitations, like occasionally a
non-selectable phenotype, or if transformats from the clone bank confer a
selectable phenotype even if they contain overexpressed wild type genes. A
second classical strategy is cloning via mapping using determination of linkage
with a large number of genetic markers on all chromosomes in meiotic tetrads.
This strategy, however, is tremendously laborious. Here, we tested if with the
advent of ever improving sequencing methods, resequencing of whole yeast
genomes has become a serious alternative method to isolate and confirm the gene
in which a given dominant mutation resides.
The
genome of both, the parent strain and the mutant derived from it, were
sequenced using the Roche/454 system. At an average read length of 499bp, this
yielded a 38.8 fold average total coverage of the genome, and divided into
19.2X coverage for K6001 and 19.5X coverage for K6001-B7.Compared to the
S288c reference, we detected 7660 uniform small nucleotide variations. 7499 of
these, proven by the very stringent criteria of a >10X coverage are
available in the Supplementary material. This rate indicates one nucleotide
exchange per 1600 bp, which points to a high degree of evolutionary divergence
between S288c and W303/K6001. Interestingly, the rate of divergence differed
between the individual chromosomes (Supplementary Figure 2).
An
interesting observation of this study was the identification of many
non-uniform variants in the haploid genome. Many of these were called with high
coverage and confidence, indicating that they might be biologically relevant
and not the result of technical artefacts. 62.1 % of the variants detected at a
calling rate between 25 and 75% were located in or close to the telomeric
regions; however, they clearly were located in unique sequences. We will
intensively discuss the nature of these variants in a future publication.
In the last three years, next generation
sequencing has been used for mapping of epigenetic mutations [19],
identification of spontaneous mutations [20], or for
evolutionary considerations [21] in yeast.
The accuracy of next generation technologies is key to this approach and in the
course of this study we identified less than 1 difference (0.86) in 1,000,000
nucleotides between both genome sequences. Lynch et al. [20] used a
pyrosequencing approach and reported an average depth of sequence coverage of
5X and restricted their analyses to sites within each genome with at least 3X
coverage. The authors pointed out that especially for homopolymeric sites, a
higher coverage depth is necessary. Recently, Araya et al. [22] used a
short read sequencing-by-synthesis approach for their whole genome sequencing
of a laboratory-evolved yeast strain. At an average depth of 28X, they covered
93% of the yeast genome. The coverage required for an in-depth analysis of
individual genomes highly depends on the used sequencing technology. According
to Wheeler et al. [23] sequencing
of diploid organisms demonstrated a minimum of 15X coverage for pyrosequencing,
and a 30X for sequencing-by-synthesis for accurate detection of heterozygous
variants. For haploid genomes, the minimal coverage depth required is lower. In
this study we reached a calling rate of 98% of the genome with an average
coverage of 19.4X for both strains. For the detection of single nucleotide
variations which distinguish K6001 from the reference genome, we merged the
genomes of both K6001 and K6001-B7, which resulted in a 38.8x fold average
genome coverage.
The
number of detected variants which distinguished our strain from the Reference
genome (12,482) was substantially large. To delimit candidate genes to identify
the mutation causing the phenotype observed, we used a rigorous subtraction
strategy. Surprisingly, the 12,482 SNV contained 5269 non-uniform variations
which had a calling rate < 95 %. We excluded these non-uniform SNVs, because
such a frequency would be incompatible with the observed 2:2 segregation of the
B7 mutant. Next, we removed all variations that were shared between K6001 and
K6001-B7, and all differences to the S288c genome that were not present in the
K6001 genome sequence. At the end, we curated the final list manually to
identify alignment artefacts of the mapping algorithm. We ended with a list of
13 differences, of which 6 were located in protein coding regions. Four of
these mutations were predicted to cause residue exchanges.
Therefore,
EMS mutagenesis had caused one mutation per ~950,000 nucleotides in the genome,
and thus, indicated that the applied protocol was efficient to create a
monogenic trait. However, we have to consider that mutant B7 was isolated after
strong selection on CHP, and therefore the unbiased primary mutation rate
immediately after EMS mutagenesis could have been substantially different.
We
started the experimental verification strategy focusing on the four residue
exchanging mutants. This choice was hypothesis driven; at this stage of the
project, also non-exonic sequences were potential, although less probable,
candidates for the B7 gene. We amplified CLC1, DON1, HOT1
and TSA1 genes by PCR from both K6001 and K6001-B7. Sanger sequencing
verified the B7-specific mutations in DON1, HOT1, and TSA1;
the six nucleotide insertion (relative to S288c) into CLC1 was also
real, but found in the parent strain as well. These four genes were then
subcloned with their native promoters into a single-copy yeast expression
vector, transformed into strain K6001, and transformants tested for their
phenotype on CHP containing media. These experiments identified and confirmed TSA1-B7as the phenotype-causing gene.
Tsa1p
(t
hiol s
pecific a
ntioxidant), a 2-Cys
peroxiredoxin, is an important enzyme of the cellular antioxidative machinery
and catalyzes H2O2 reduction in the presence of
thioredoxin, thioredoxin reductase and NADPH [24]. Tsa1p
furthermore acts as a molecular chaperone, and is associated with ribosomes, it
prevents oxidative damage of newly synthesized polypeptides [25]. It was
previously shown that disruption of Tsa1p diminishes oxidative stress
resistance of yeast; cells lacking this enzyme were highly sensitive to
tert-butyl hydroperoxide, hydrogen peroxide, and CHP [26].
Peroxiredoxin 1 has been associated to the aging of mammals, since it interacts
and stimulates the activity of the lifespan regulator protein p66Shc [27], and
disruption of Tsa1p in yeast shortens its chronological lifespan [28]. The
results presented here show that the involvement of Tsa1 in aging phenotype is
complex. In contrast to the TSA1 knock-out, yeast cells expressing the TSA1-B7
allele gain resistance to oxidants, but are also compromised in a lifespan
phenotype. Thus, changes in the natural antioxidative capacities of the
peroxiredoxin system in either direction can accelerate yeast aging. These
results add and substantiate other observations that highlight the importance
of a natural redox balance, rather than the total antioxidative capacity, as an
important determinant of cellular lifespan [29-31]. For
instance, C. elegans requires a natural rate of free radical generation
for lifespan extension by caloric restriction [32], and yeast
cells, which are oxidant-resistant due to an excessive NADPH production caused
by mutations in triose phosphate isomerase gene, are also replicatively short
lived [8]. Thus,
understanding the influence of free radicals and oxidative stress on the
complex phenotype of aging requires examination of these processes in the
context of the highly evolved regulation of the cellular redox environment.
Our
study demonstrates that whole-genome re-sequencing is suitable to identify a
functional single nucleotide exchange generated by random mutagenesis. Although
all commercially available sequencing platforms (Genome Analyzer (Illlumina),
SOLiD (Life Technologies) and FLX Genome Sequencer (454/Roche)) would provide
appropriate workflows, we decided on 454 sequencing because of large
read-lengths, sequence accuracy and the available software tools. All data was
collected in a single Titanium run on the FLX sequencer. Thus, whole genome
re-sequencing strategies have the potential to increase the efficiency and
flexibility of random strategies, highly increasing their attractiveness for
addressing current biological problems.
Materials and Methods
Yeast cultivation and mutagenesis.
Yeast was
cultivated on yeastextract peptone dextrose (YPD) or galactose (YPGal),
synthetic complete glucose (SC) or galactose (SCGal) media at 28°C. For EMS
mutagenesis, logarithmically growing K6001
cells were washed twice with 50 mM potassium phosphate (pH 6.8), and resuspended
in 10 ml of this buffer supplemented with 300μl EMS. After one hour incubation
at 28°C, where 10% of the cells were still alive, mutagenesis was stopped by
adding 10ml 10% w/v sodium thiosulfate [33].
qRT-PCR.
Yeast total RNA was extracted using
RiboPure-Yeast Kit (Ambion). After quality control, cDNA was synthezised using
12-18 oligo dT primers and Moloney Murine Leukemia virus (M-MuLV)
reverse transcriptase (NEB) according to the manufacturer's instructions.
Real-time PCRs were performed in triplicates in a final volume of 5 μl
containing 1 μl cDNA, 1 μl 5x combinatorial enhancer solution (CES) [34], 0.5 μl
primer mix and 2.5 μl 2× SybrGreen master mix (Fermentas). Reactions were run
on a Prism 7900HT sequence detection system (ABI). The thermal cycling
conditions comprised 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for
15 s/60°C for 1 min. The relative expression ratio of the target gene TSA1
was normalized to the geometric mean of three endogenous reference transcripts
(ACN9, ATG27, TAF10) by the method of Pfaffl [35].
Cloning
and sanger sequencing.
Candidate
genes were amplified by oligonucleotides given in table 1 using Phusion
polymerase (Finnzymes). The resulting products were gel-purified and used a)
for Sanger resequencing and b) subcloned into a pRS413-derived yeast
single-copy centromeric expression vector. For this, the products were treated
with the endonucleases (New England Biolabs) given in Table 1, and ligated into
compatible sites of the yeast vector. 2μ overexpression plasmids p423-TSA1
and p423-TSA1-B7 were generated by excision of the corresponding
fragments with SacI/XhoI from the cen-plasmid, and their ligation
into the backbone of the SacI/XhoI digested 2μ vector p423GPD [36]. Plasmids
were verified by endonuclease digestion and sequencing.
Targeted
protein quantification by mass spectrometry.
Tsa1p levels were quantified by the means of determination of their
relative abundance relative to the expression of triosephosphate isomerase
(Tpi1p). Whole-proteome tryptic digestes were generated and analyzed by
nanoflow liquid chromatography tandem mass spectrometry (nanoLC-MS/MS) on an
QTRAP5500 hybrid triple quadrupole/ion trap mass spectrometer (AB/Sciex) as
described earlier [37]. Analyzed
peptides and the MRM transitions used for quantification are given in Table 2.
Lifespan
assay by micromanipulation.
Logarithmically
growing yeast cultures were plated at low density, and at least 50 daughter
cells were isolated as buds with a MSM micromanipulator (Singer instruments).
After their first division, mothers were removed and 2nd generation
virgin cells kept for analysis. The lifespan of these cells was determined by
counting and removing all subsequent daughters at 28°C. Cells were shifted to
8°C overnight to allow resting of the investigator; depending on the age of the
cells, 1-2 generations were completed at this temperature per night. Statistical
calculations for lifespans were conducted with SPSS 11.0 (SPSS) and Excel with
Winstat. By applying Kaplan-Meier statistics the standard deviations of the
median lifespan at a confidence level of 95% were calculated. To show if two
given survival distributions are significantly different at a 95% confidence
level, logrank (Mantel-Cox), the modified Wilcoxon test statistic (Breslow),
and Tarone & Ware statistics were used as described earlier [16].
454
Sequencing and data analysis.
DNA was
sheared by sonication to a fragment size of 500 - 800bp, and adaptors ligated.
The amplified template beads were recovered after emulsion breaking and
selective enrichment. Sequencing primer was annealed to the template and the
beads were incubated with Bst DNA polymerase, apyrase, and
single-stranded binding protein. Template beads, enzyme beads (required for
signal transduction) and packing beads (for Bst DNA polymerase
retention) were loaded into the wells of a 70 x 75 mm two compartment picotiter
plate. The picotiter plate was inserted in the flow cell and subjected to
pyrosequencing on the Genome Sequencer FLX instrument (454/Roche).
The system flows 200 cycles of four solutions
containing either dTTP, alphaSdATP, dCTP or dGTP reagents, in that order, over
the cell. For each dNTP flow, a single image was captured by a charge-coupled
device (CCD) camera on the sequencer. The images were processed to identify DNA
bead-containing wells and to compute associated signal intensities. The images
were further processed for chemical and optical cross-talk, phase errors, and
read quality before base calling was performed for each template bead.
Raw data processing.
After default raw data processing, we used a
resequencing trimming filter to increase the data output. (parameters doValleyFilterTrimBack
= false, vfBadFlowThreshold = 6, vfLastFlowToTest = 168, errorQscoreWindowTrim
= 0.01).
Mapping 454 reads to a reference
genome.
We generated 1.3 million
sequences which produced 662 million bases that were aligned to the Saccharomyces
cerevisiae reference genome [38], using GS
reference mapper version 2.3. The best match in the genome was used as the
location for the reads with multiple matches. SNPs and small
insertion-deletions were included in the analysis.
Filtering
small nucleotide variations.
To
identify the phenotype-causing SNP of the B7, we first removed non-uniform SNVs
and insertions/deletions by introducing a cutoff-filter of <95%. Next, we
eliminate those variants which were equal in B7 and K6001. The remaining 152
differences were controlled by hand, taking into account the covered position
of the yeast genome, the reference base and the consensus base for each
position (‘454AlignmentInfo' file). Of these, we manually compared all variants
called with at least three non-duplicate reads in both directions with the
consensus base position in K6001.
Acknowledgments
We thank Beata Lukaszewska-McGreal for help with sample
preparation and our lab-members for critical discussion. We are grateful to the
EC (Brussels, Europe) for project MIMAGE (contract no. 512020; to M.B.) and to
the Austrian Science Fund FWF (Vienna, Austria) for grant S9302-B05 (to M.B.)
and S9306 (to JG).
Conflicts of Interest
The authors of this manuscript have no conflict of
interests to declare.
References
-
1.
Finkel
T
and Holbrook
NJ.
Oxidants, oxidative stress and the biology of ageing.
Nature.
2000;
408:
239
-247.
[PubMed]
.
-
2.
Muller
FL
, Lustgarten
MS
, Jang
Y
, Richardson
A
and Van
Remmen H.
Trends in oxidative aging theories.
Free Radic Biol Med.
2007;
43:
477
-503.
[PubMed]
.
-
3.
Harman
D
Aging: a theory based on free radical and radiation chemistry.
J Gerontol.
1956;
11:
298
-300.
[PubMed]
.
-
4.
Aguilaniu
H
, Gustafsson
L
, Rigoulet
M
and Nystrom
T.
Asymmetric inheritance of oxidatively damaged proteins during cytokinesis.
Science.
2003;
299:
1751
-1753.
[PubMed]
.
-
5.
Klinger
H
, Rinnerthaler
M
, Lam
YT
, Laun
P
, Heeren
G
, Klocker
A
, Simon-Nobbe
B
, Dickinson
JR
, Dawes
IW
and Breitenbach
M.
Quantitation of (a)symmetric inheritance of functional and of oxidatively damaged mitochondrial aconitase in the cell division of old yeast mother cells.
Exp Gerontol.
2010;
45:
533
-542.
[PubMed]
.
-
6.
Blagosklonny
MV
Aging: ROS or TOR.
Cell Cycle.
2008;
7:
3344
-3354.
[PubMed]
.
-
7.
Heeren
G
, Rinnerthaler
M
, Laun
P
, von
Seyerl P
, Kössler
S
, Klinger
H
, Hager
M
, Bogengruber
E
, Jarolim
S
, Simon-Nobbe
B
, Schüller
C
, Carmona-Gutierrez
D
and Breitenbach-Koller
L.
The mitochondrial ribosomal protein of the large subunit, Afo1p, determines cellular longevity through mitochondrial back-signaling via TOR1.
Aging.
2009;
1:
622
-636.
[PubMed]
.
-
8.
Ralser
M
, Wamelink
MM
, Kowald
A
, Gerisch
B
, Heeren
G
, Struys
EA
, Klipp
E
, Jakobs
C
, Breitenbach
M
, Lehrach
H
and Krobitsch
S.
Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress.
J Biol.
2007;
6:
10
[PubMed]
.
-
9.
Sanz
A
, Fernandez-Ayala
DJ
, Stefanatos
RK
and Jacobs
HT.
Mitochondrial ROS production correlates with, but does not directly regulate lifespan in Drosophila.
Aging.
2010;
2:
220
-223.
[PubMed]
.
-
10.
Postma
L
, Lehrach
H
and Ralser
M.
Surviving in the cold: yeast mutants with extended hibernating lifespan are oxidant sensitive.
Aging.
2009;
1:
957
-960.
[PubMed]
.
-
11.
Blagosklonny
MV
and Hall
MN.
Growth and aging: a common molecular mechanism.
Aging.
2009;
1:
357
-362.
[PubMed]
.
-
12.
Winzeler
EA
, Shoemaker
DD
, Astromoff
A
, Liang
H
, Anderson
K
, Andre
B
, Bangham
R
, Benito
R
, Boeke
JD
, Bussey
H
, Chu
AM
, Connelly
C
and Davis
K.
Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis.
Science.
1999;
285:
901
-906.
[PubMed]
.
-
13.
Powers
RW 3rd
, Kaeberlein
M
, Caldwell
SD
, Kennedy
BK
and Fields
S.
Extension of chronological life span in yeast by decreased TOR pathway signaling.
Genes Dev.
2006;
20:
174
-184.
[PubMed]
.
-
14.
Kaeberlein
M
, Powers
RW 3rd
, Steffen
KK
, Westman
EA
, Hu
D
, Dang
N
, Kerr
EO
, Kirkland
KT
, Fields
S
and Kennedy
BK.
Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients.
Science.
2005;
310:
1193
-1196.
[PubMed]
.
-
15.
Bobola
N
, Jansen
RP
, Shin
TH
and Nasmyth
K.
Asymmetric accumulation of Ash1p in postanaphase nuclei depends on a myosin and restricts yeast mating-type switching to mother cells.
Cell.
1996;
84:
699
-709.
[PubMed]
.
-
16.
Jarolim
S
, Millen
J
, Heeren
G
, Laun
P
, Goldfarb
DS
and Breitenbach
M.
A novel assay for replicative lifespan in Saccharomyces cerevisiae.
FEMS Yeast Res.
2004;
5:
169
-177.
[PubMed]
.
-
17.
Laun
P
, Pichova
A
, Madeo
F
, Fuchs
J
, Ellinger
A
, Kohlwein
S
, Dawes
I
, Frohlich
KU
and Breitenbach
M.
Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis.
Mol Microbiol.
2001;
39:
1166
-1173.
[PubMed]
.
-
18.
Breitenbach
M
, Dickinson
R
and Laun
P.
Smart Genetic Screens. In Methods in Microbiology. Volume 36.
Edited by Stansfield I, Stark M: Academic Press;.
2007;
331
-367.
.
-
19.
Irvine
DV
, Goto
DB
, Vaughn
MW
, Nakaseko
Y
, McCombie
WR
, Yanagida
M
and Martienssen
R.
Mapping epigenetic mutations in fission yeast using whole-genome next-generation sequencing.
Genome Res.
2009;
19:
1077
-1083.
[PubMed]
.
-
20.
Lynch
M
, Sung
W
, Morris
K
, Coffey
N
, Landry
CR
, Dopman
EB
, Dickinson
WJ
, Okamoto
K
, Kulkarni
S
, Hartl
DL
and Thomas
WK.
A genome-wide view of the spectrum of spontaneous mutations in yeast.
Proc Natl Acad Sci U S A.
2008;
105:
9272
-9277.
[PubMed]
.
-
21.
Gresham
D
, Desai
MM
, Tucker
CM
, Jenq
HT
, Pai
DA
, Ward
A
, DeSevo
CG
, Botstein
D
and Dunham
MJ.
The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast.
PLoS Genet.
2008;
4:
e1000303
[PubMed]
.
-
22.
Araya
CL
, Payen
C
, Dunham
MJ
and Fields
S.
Whole-genome sequencing of a laboratory-evolved yeast strain.
BMC Genomics.
2010;
11:
88
[PubMed]
.
-
23.
Wheeler
DA
, Srinivasan
M
, Egholm
M
, Shen
Y
, Chen
L
, McGuire
A
, He
W
, Chen
YJ
, Makhijani
V
, Roth
GT
, Gomes
X
, Tartaro
K
and Niazi
F.
The complete genome of an individual by massively parallel DNA sequencing.
Nature.
2008;
452:
872
-876.
[PubMed]
.
-
24.
Wood
ZA
, Schröder
E
, Robin
Harris J
and Poole
LB.
Structure, mechanism and regulation of peroxiredoxins.
Trends Biochem Sci.
2003;
28:
32
-40.
[PubMed]
.
-
25.
Trotter
EW
, Rand
JD
, Vickerstaff
J
and Grant
CM.
The yeast Tsa1 peroxiredoxin is a ribosome-associated antioxidant.
Biochem J.
2008;
412:
73
-80.
[PubMed]
.
-
26.
Chae
HZ
, Kim
IH
, Kim
K
and Rhee
SG.
Cloning, sequencing, and mutation of thiol-specific antioxidant gene of Saccharomyces cerevisiae.
J Biol Chem.
1993;
268:
16815
-16821.
[PubMed]
.
-
27.
Gertz
M
, Fischer
F
, Leipelt
M
, Wolters
D
and Steegborn
C.
Identification of Peroxiredoxin 1 as a novel interaction partner for the lifespan regulator protein p66Shc.
Aging.
2009;
1:
254
-265.
[PubMed]
.
-
28.
Lee
JH
and Park
J-W.
Role of Thioredoxin Peroxidase in Aging of Stationary Cultures of Saccharomyces cerevisiae.
Free Radical Research.
2004;
38:
225
-231.
[PubMed]
.
-
29.
Zuin
A
, Castellano-Esteve
D
, Ayte
J
and Hidalgo
E.
Living on the edge: stress and activation of stress responses promote lifespan extension.
Aging.
2010;
2:
231
-237.
[PubMed]
.
-
30.
Ralser
M
and Benjamin
IJ.
Reductive stress on life span extension in C. elegans.
BMC Res Notes.
2008;
1:
19
[PubMed]
.
-
31.
Rattan
SI
Hormesis in aging.
Ageing Res Rev.
2008;
7:
63
-78.
[PubMed]
.
-
32.
Schulz
TJ
, Zarse
K
, Voigt
A
, Urban
N
, Birringer
M
and Ristow
M.
Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress.
Cell Metab.
2007;
6:
280
-293.
[PubMed]
.
-
33.
Burke
D
, Dawson
D
and Stearns
T.
Plainview, N.Y.
Cold Spring Harbor Laboratory Press
Cold Spring Harbor Laboratory. Methods in yeast genetics : a Cold Spring Harbor Laboratory course manual. 2000 edn.
2000;
.
-
34.
Ralser
M
, Querfurth
R
, Warnatz
HJ
, Lehrach
H
, Yaspo
ML
and Krobitsch
S.
An efficient and economic enhancer mix for PCR.
Biochem Biophys Res Commun.
2006;
347:
747
-751.
[PubMed]
.
-
35.
Pfaffl
MW
A new mathematical model for relative quantification in real-time RT-PCR.
Nucleic Acids Res.
2001;
29:
e45
[PubMed]
.
-
36.
Mumberg
D
, Muller
R
and Funk
M.
Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds.
Gene.
1995;
156:
119
-122.
[PubMed]
.
-
37.
Wamelink
MM
, Gruning
NM
, Jansen
EE
, Bluemlein
K
, Lehrach
H
, Jakobs
C
and Ralser
M.
The difference between rare and exceptionally rare: molecular characterization of ribose 5-phosphate isomerase deficiency.
J Mol Med.
2010;
.
-
38.
Goffeau
A
, Barrell
BG
, Bussey
H
, Davis
RW
, Dujon
B
, Feldmann
H
, Galibert
F
, Hoheisel
JD
, Jacq
C
, Johnston
M
, Louis
EJ
, Mewes
HW
and Murakami
Y.
Life with 6000 genes.
Science.
1996;
274:
546, 563
-547.
[PubMed]
.