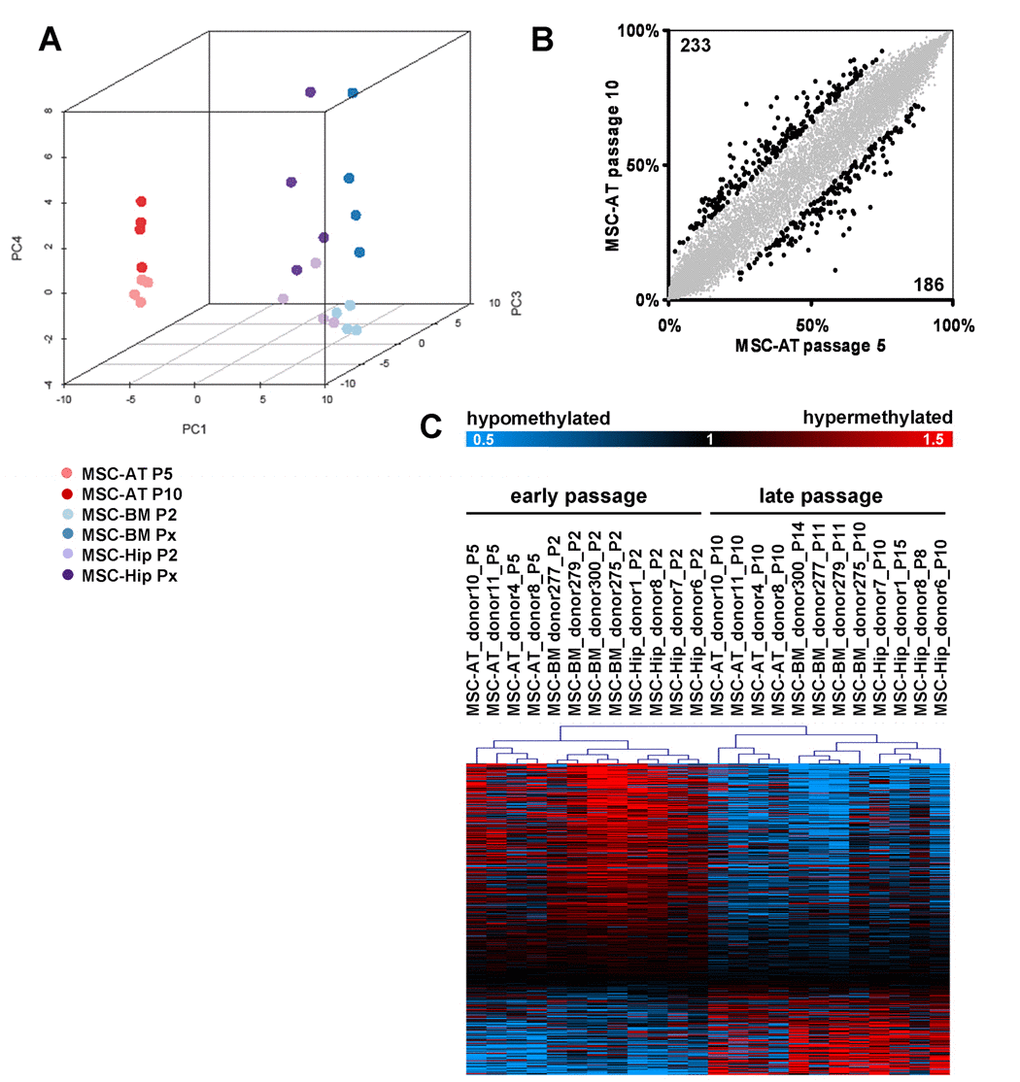
Figure 3.Senescence-associated modifications in the DNA-methylation patternDNA-methylation profiles were analyzed with the HumanMethylation27 BeadChip microarray which represents 27,578 unique CpG sites. MSC derived from adipose tissue (MSC-AT) were compared with those derived from bone marrow, which was either aspirated from the iliac crest (MSC-BM) or taken from the caput femuris upon hip replacement (MSC-Hip). Unsupervised principal component analysis (PCA) clearly separated DNA-methylation profiles according to the tissue of origin in the first dimension (PC1), whereas the forth component (PC4) discerned early and late passage (A). Scatterplot comparison of passage 5 and passage 10 in MSC-AT revealed that 233 CpG sites are more than 15% hyper-methylated whereas 186 CpG sites are more than 15% hypo-methylated at passage 10 (B). Significance Analysis of Microarray (SAM) was used to select 517 senescence-associated CpG sites (FDR = 4.8%) and these are presented as a heatmap (C; data were divided by the mean of each row for graphical presentation).