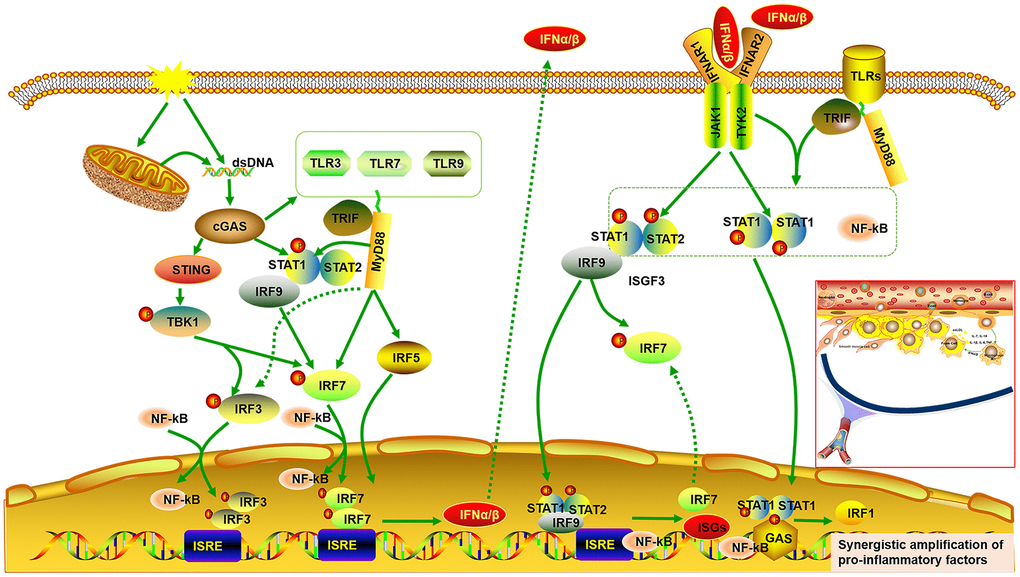
Figure 9.Signal integrations of TLRs, STAT/IRF as well as type-I IFN exacerbate synergistic amplification of gene expression that leads to an inflammatory cascade and pro-atherogenic responses. cGAS is activated by dsDNA and triggers the downstream TANK binding kinase (TBK)1, followed by phosphorylation of IRF3 as well as IRF7, which form homodimer and enter the nucleus from cytoplasm, accompany with or without other transcription factors such as STAT and NF-kB, thereby allowing initiation of the subsequent production of type I IFNα/β. On the other hand, the binding of type I IFN to the IFNα/β receptor (IFNAR)2 recruits IFNAR1. This complex enables activation of the receptor-associated JAK1 and tyrosine kinase (TYK)2, followed by STAT1 and STAT2 phosphorylation, which bind to IRF9, forming IFN-stimulated gene factor (ISGF)3 complex. The ISGF3 complex translocates into nucleus and promote the production of ISGs and IRF7 by binding to IFN-stimulated regulatory elements (ISRE) as well as IRF7 elements in DNA. Phosphorylated STAT1 can also form a homodimer, which binds to a comparable γ-activated sequence (GAS) in DNA, inducing the expression of IRF1 and pro-inflammatory genes. Moreover, cGAS can also result in up-regulated expression of TLRs (TLR3, TLR7, TLR9) and STAT (STAT1, STAT2). STAT1 phosphorylation can be induced by multiple TLRs dependent on MyD88 and TRIF signaling. Phosphorylated STAT1 translocates into the nucleus and augments TLR-NF-kB activation, promoting the expression of pro-inflammatory genes. Multiple TLRs also activate IRF5 and IRF7 as well as IRF3 via MyD88 and TRIF signaling.