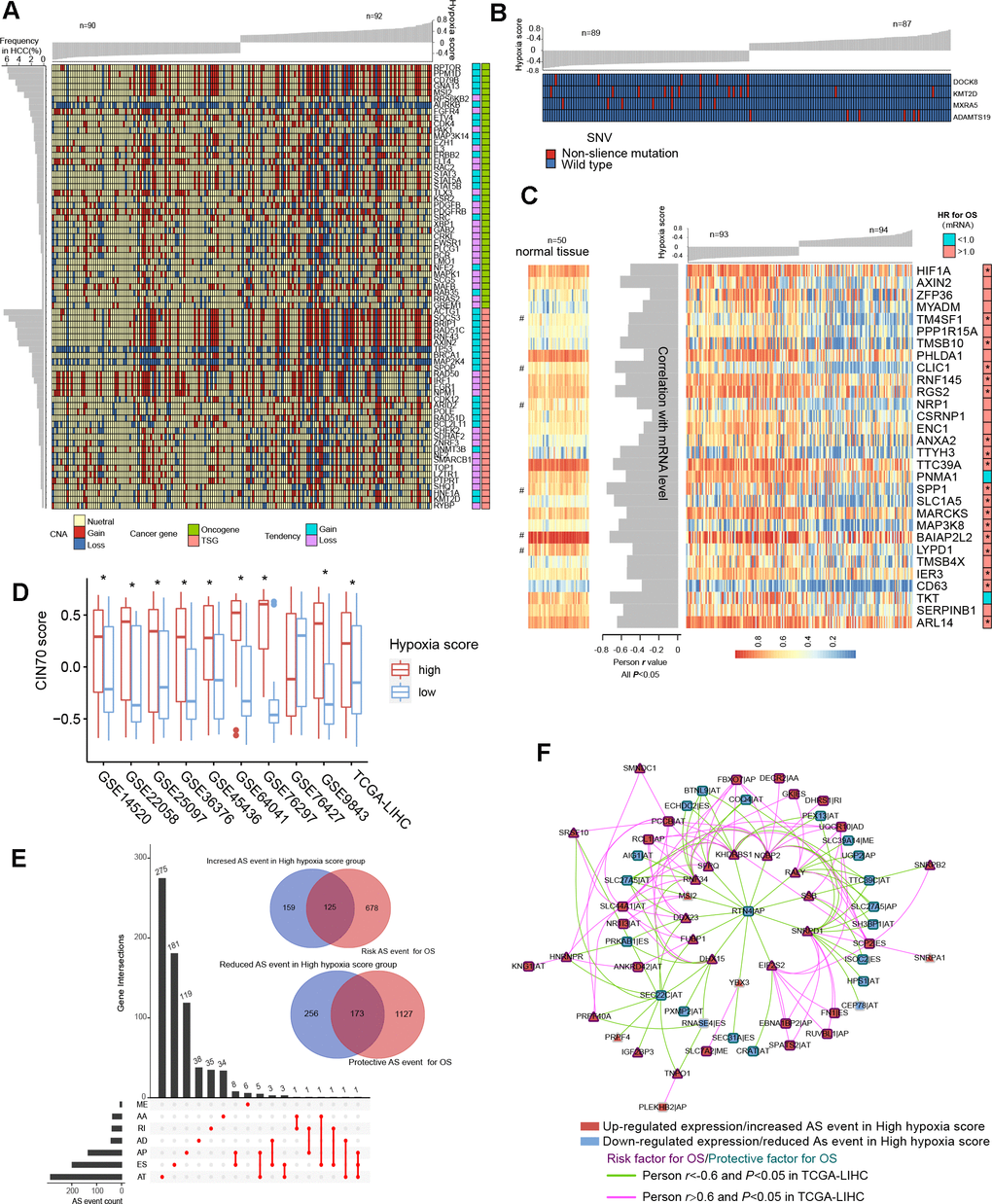
Figure 4.Differences in genomic and epigenetic alterations between groups with high hypoxia scores and low hypoxia scores. (A) The difference in the incidence of copy-number aberrations (CNAs) in 71 cancer genes between the high hypoxia score group and the low hypoxia score group. (B) The proportions of single-nucleotide variants (SNVs) in 4 genes are significantly different between the high hypoxia score group and the low hypoxia score group. (C) Reductions in the methylation levels of 30 genes are accompanied by significant increases in the corresponding mRNA levels in the high hypoxia score group. The correlation between DNA methylation and corresponding mRNA expression was obtained through Pearson correlation analysis based on TCGA-LIHC data. The hazard ratios (HR) of the corresponding mRNAs for overall survival (OS) were calculated by the logrank test for TCGA-LIHC data, and the cut-off was the median expression level. (D) 70-gene chromosome instability (CIN70) was used to assess chromosome instability in tumor tissues from 10 hepatocellular carcinomas (HCC) datasets. The CIN70 scores are significantly different between tumor tissues with high hypoxia scores and low hypoxia scores. (E) The occurrences of 713 AS events are significantly different between the high hypoxia score group and the low hypoxia score group. Some of the AS events are associated with the OS of HCC patients. (F) The expression of 30 splicing factors in the high hypoxia score group and the low hypoxia score group are different, and their expression trends are consistent in 10 datasets. These splicing factors and the AS events with different occurrences between the two groups form a network. The correlations between the nodes of the network were calculated by Pearson correlation analysis based on TCGA-LIHC data. The relationship between the nodes and the OS of HCC patients was obtained through univariate cox survival analysis.