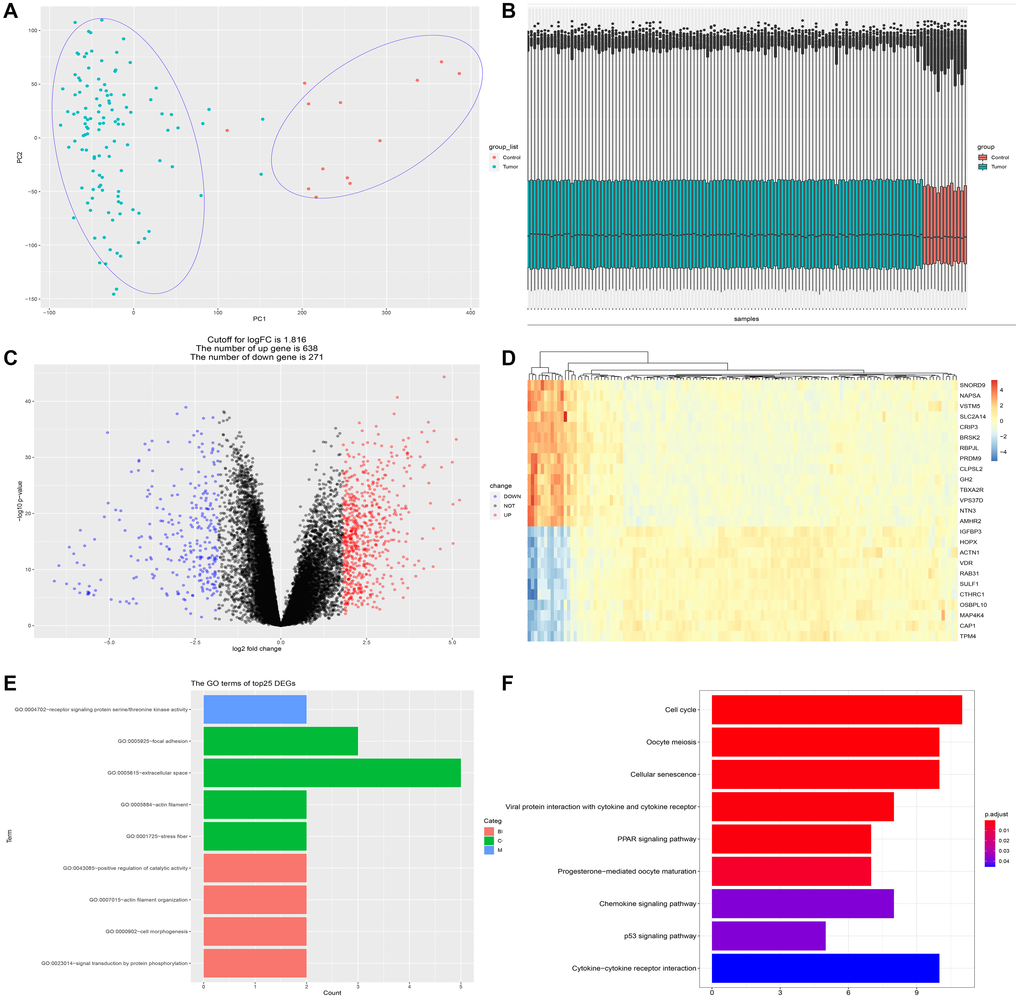
Figure 1.Identification of differentially expressed genes (DEGs) between PDAC and normal pancreatic tissue. (A) PCA analysis discriminates PDAC group (n = 118) from control group (n = 13). (B) The dataset (GSE62165) conformed to sample homogeneity. (C) A total of 909 DEGs were obtained in GSE62165, among which 271 genes were down-regulated and 638 genes up-regulated. (D) The top 25 DEGs exhibited by heatmap. (E) The GO terms of the top 25 DEGs. GO analysis of top 25 DEGs is related to cell skeleton components (extracellular space, actin filament, stress fiber, etc.) and protein phosphorylation events. (F) The KEGG analysis of the top 25 DEGs. The most significant pathways are cell cycle regulation, cellular senescence, PPAR signaling, P53 signaling, chemokine signaling and cytokine-cytokine receptor interaction.