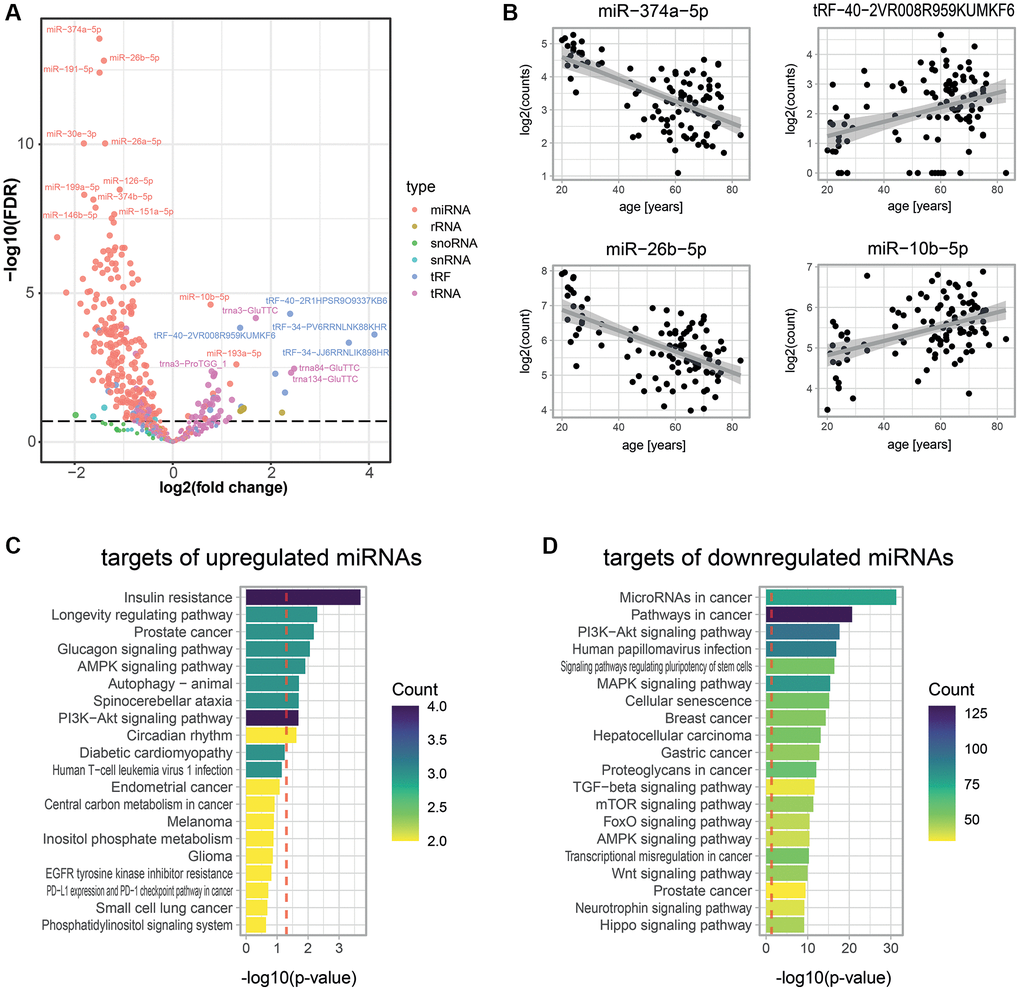
Figure 3.Age-associated small RNAs in blood plasma. (A) Age-dependent changes for 608 small RNAs as measured by Small-seq in 103 individuals. A negative binomial model was fitted for each RNA using DESeq2 [79]. The volcano plot shows log2 fold changes in expression between young and old individuals on the x-axis and log10 p-values of BH FDR-corrected Wald tests on the y-axis. The former was obtained by multiplying the log2 fold change in small RNA expression for 1 year (i.e., the estimate of the model) with the mean age difference between individuals from the young and old age groups of the untargeted proteomics experiments (i.e., 44.8 years). (B) Examples of scatter plots for four small RNAs detected as age-associated. (C, D) KEGG pathway enrichment analysis for predicted targets of significantly up- (C) and down- (D) regulated miRNAs. A robust analytic approach (see Methods) allowed us to select 22 and 2,159 miRNA targets that were up- and down-regulated with age, respectively. The set of all 26,194 human transcripts present in the multiMIR database was used as a background to compute the significance of age-association. The gprofiler2 R package was used to compute enrichment, and p-values were corrected using the gSCS correction method [85]. Colors show the number of targets of our age-associated miRNAs that are attributed to these pathways.
Figure 3 — Age prediction from human blood plasma using proteomic and small RNA data: a comparative analysis | Aging