



Introduction
Alzheimer's disease (AD) is an aging-related progressive neurodegenerative disease, characterized by massive neuronal and synaptic loss, accompanied by neuropathological changes, such as neurofibrillary tangles and senile plaques, in the hippocampus, neocortex, and subcortical structures [1]. The senile plaques are primarily composed of amyloid beta peptide (Aβ), which is a 40-42 amino acid peptide fragment of the amyloid protein precursor. However, the mechanism by which Aβ causes neuronal injury and cognitive impairment is unclear. AD is also thought to have a local, non-immune mediated neuroinflammatory component with clusters of activated microglia, increased inflammatory proteins (complement factors, acute-phase protein, pro-inflammatory cytokines) [2-4], and increased COX-1-expressing microglia surrounding amyloid plaques [2]. Changes in COX-2 expression in AD are discrepant and seem to depend on the stage of the disease, with an upregulation of COX-2 in early AD, and a downregulation in advanced AD stages, which also correlate with PGE2 levels in the CSF, which are increased in probable AD patients and decrease with the progression of the disease [5,6]. Several independent epidemiological studies have shown that early use of non steroidal anti-inflammatory drugs (NSAIDs), which inhibit COX activity, significantly reduces the risk of developing AD later in life suggesting that inflammation is critical for the progression of the disease [7-13]. However, although a 6-month, double-blinded, placebo-controlled study with indomethacin, a preferential COX-1 inhibitor, appeared to protect AD patients from cognitive decline [14], subsequent large-scale randomized clinical trials, mostly with selective COX-2 inhibitors, did not show any beneficial effects in AD patients with mild to severe symptoms [15-18]. Supporting these clinical data, indomethacin, but not the COX-2 selective nimesulide, significantly reduced levels of Aβ in the hippocampus and cortex of transgenic mouse models of AD [19]. While the clinical data seem to rule out a protective effect of selective COX-2 inhibition in AD, it is still unclear whether COX-2 inhibitors can improve the pathology in animal models of AD. For instance, COX-2 inhibition blocks Aβ-mediated suppression of long-term potentiation and memory function, independently of reductions in Aβ1-42 or in inflammation [20]. However, the selective COX-2 inhibitor celecoxib has been shown to increase Aβ levels [21,22], and in a model of acute inflammation, both genetic deletion and pharmacological inhibition of COX-2 worsen the neuroinflammatory response to lipopolysaccharide (LPS) [23]. These combined data suggest that either NSAIDs have rather a preventive than a therapeutic effect or that preferential COX-1 inhibition is a better therapeutic approach than selective targeting COX-2, or that the beneficial effects are due to COX-independent effects of NSAIDs. In particular, ibuprofen, flurbiprofen, and diclofenac have been shown to reduce serum Aβ1-42 levels, a major component of senile plaques in AD [24-28]. However, a recent report from a pooled dataset from six prospective studies indicated that NSAIDs use reduced the risk of AD without any apparent advantage for the subset of NSAIDs shown to selectively lowering Aβ1-42[29]. While COX-1 and COX-2 are both differentially expressed in different stages of AD pathology, their specific roles in the pathogenesis of AD is unclear. Therefore, a full understanding of the physiological, pathological, and/or neuroprotective role of COX isoforms may help to develop better therapeutic strategies for the prevention or treatment of AD.
Partial reproduction of AD neuropathology and cognitive deficits has been achieved with pharma-cological and genetic approaches. Most injection models use synthetic peptide Aβ1-40 or Aβ1-42, which are analogous to peptides found in neuritic plaques in AD patients [30]. Mice with a null mutation of COX gene have been a useful tool for investigating the role of each COX isoform in both physiological and pathological conditions in the CNS by overcoming the complexity of dosing paradigm, duration of treatment, and possible unspecific inhibition of both COX isoform [31]. In this study, we assessed the effect of intracerebroventricular (i.c.v.) injection of Aβ1-42 on acute neuroinflammatory response in COX-1-deficient (COX-1-/-) mice and their respective wild-type mice (WT) controls. We showed that COX-1-/- mice are more resistant than WT mice to Aβ1-42-induced neuronal death and exhibit a marked reduction in the inflammatory response.
Results
The inflammatory response is reduced in COX-1-/- mice after Aβ1-42 injection
Aβ1-42 or the control reverse peptide Aβ42-1 was unilaterally injected into the lateral ventricle, as reported [32-35]. Seven days later, brains were removed and coronal sections were processed for immunohistochemistry. We assessed microglial activation in the brain using IBA-1 as a microglial marker. Aβ1-42 administration caused a robust inflammatory response within the CA1 and CA3 areas of the hippocampus of WT mice characterized primarily by the presence of activated microglia (Figure 1A, D, J). Intense IBA-1-immunoreactive microglia with enhanced staining intensity, retracted processes, perikaryal hypertrophy, and amoeboid appearance were observed in the CA3 area of hippocampus of WT mice (Figure 1G). In COX-1-/- mice, IBA-1-immunreactive microglia retained a resting morphology with specifically small cell bodies, thin, and ramified processes (Figure 1B, E, H, J). In reverse peptide Aβ42-1-injected mice, only a few faintly IBA-1-immunoreactive microglia were observed in the hippocampus (Figure 1C, F, I, J). Staining with CD11b, another marker for microglia gave results similar to that of IBA-1 (data not shown).
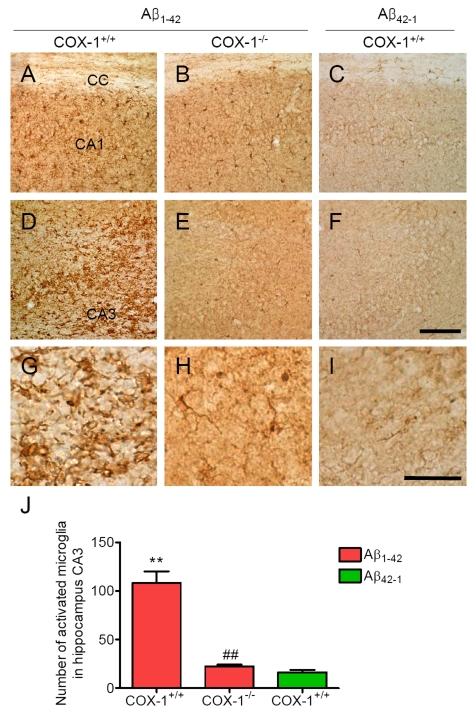
Figure 1. Increased microglial activation in the hippocampus 7 d after Aβ 1-42 administration. Representative
photomicrographs of the CA1 and CA3 of the hippocampus from WT mice (A,
D) injected with Aβ1-42
that shows numerous activated microglia with short, less-ramified
processes, perikaryal hypertrophy, and amoeboid appearance (G). CA1 and CA3 areas of the hippocampus from Aβ1-42-injected
COX-1-/- mice (B, E) show many resting microglia
with ramified morphology (H). Scale bar: A-F, 100
μm; G-I, 50 μm. (J) Comparison of the number of activated
microglia from the CA3 area. Mean ± SEM (n = 3-4 per group); **P
< 0.01 compared with the Aβ42-1-injected WT mice; ##P
< 0.01 compared with the Aβ1-42-injected WT mice.
We then assessed astrocytes immunoreactivity by staining the brain of WT and COX-1-/- mice with the astrocytic marker glial fibrillary acidic protein (GFAP). GFAP-immunoreactive astrocytes in response to Aβ1-42 injection were markedly attenuated in the brain of COX-1-/- mice (Figure 2B, E, H) compared to WT mice (Figure 2A, D, G). These results indicate that Aβ1-42 administration induced less severe glial cell activation in COX-1-/- mice compared to WT mice.
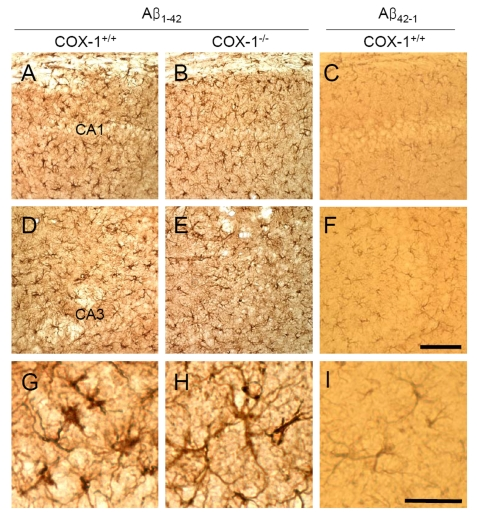
Figure 2. Increased astrocytic activation in the hippocampus 7 d after Aβ 1-42 administration. Representative
photomicrographs of the CA1 and CA3 of the hippocampus from WT mice (A,
D, G) injected with Aβ1-42 that shows numerous robustly
GFAP-immunoreactive astrocytes compared with Aβ1-42-injected
COX-1-/- mice (B, E, H). Scale
bar: A-F, 100 μm; G-I, 50 μm.
COX-1 deficiency leads to reduced neuronal damage following Aβ1-42 injection
We next assessed neuronal damage in the brain using the fluorescent marker Fluoro-Jade B (FJB), which selectively labels injured neurons [36,37]. Aβ1-42 administration caused a significant neuronal damage, characterized by the presence of FJB-positive neurons within the CA3 areas of hippocampus of WT mice (Figure 3A, J). In contrast, Aβ1-42-injected COX-1-/- mice showed few scattered FJB-positive neurons in the CA3 of hippocampus (Figure 3B, J). In same sections stained with DAPI or adjacent sections stained with cresyl violet, a similar distribution of neuronal loss and gliosis was found in the CA3 areas of hippocampus in Aβ1-42-injected WT mice (Figure 3D, G). FJB and Nissl staining showed that hippocampal CA3 neurons in COX-1-/- mice were better preserved than in WT mice (Figure 3E, H). These results indicate that Aβ1-42 administration induced less severe neuronal damage in COX-1-/- mice compared to WT mice.
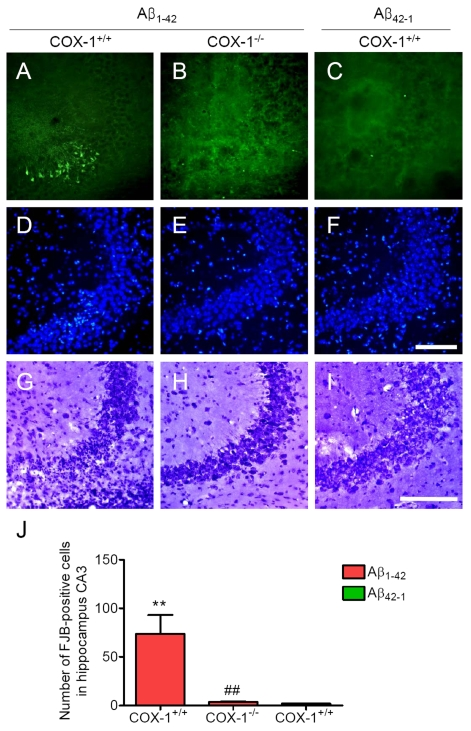
Figure 3. Increased degenerating neurons in the hippocampus 7 d after. Aβ1-42
administration. (A-C)
Representative photomicrographs of the CA3 of the hippocampus from WT mice
(A) injected with Aβ1-42 that shows numerous FJB-positive cells
compared with Aβ1-42-injected COX-1-/-
mice (B). Representative photomicro-graphs of DAPI (D-F) and
Nissl staining (G-I) in the CA3 of hippocampus from Aβ1-42-injected WT (D, G) and COX-1-/-mice (E, H).
Scale bar: A-I, 100 μm. (J) Comparison of the number of
FJB-positive cells from the CA3 area. Mean ± SEM (n = 3-4 per
group); **P < 0.01 compared with the Aβ42-1-injected
WT mice; ##P < 0.01 compared with the Aβ1-42-injected
WT mice.
COX-1-/- mice exhibit reduced oxidative damage following Aβ1-42 administration
An important component of Aβ1-42-induced neurotoxic process is mediated by oxidative damage [38], which can be evaluated by assessing protein carbonyls and nitrotyrosine levels [39]. To determine whether oxidative damage is involved in the process of Aβ1-42-induced neurotoxic process, we investigated oxidized amino acid, nitrotyrosine levels using sections adjacent to those used for FJB staining. We found an increase in nitrotyrosine-immunoreactive cells in the brain of WT mice (Figure 4A, D), which was markedly attenuated in the brain of COX-1-/- mice (Figure 4B, E). These results indicate that Aβ1-42 administration induced less severe oxidative damage in COX-1-/- mice compared to WT mice.
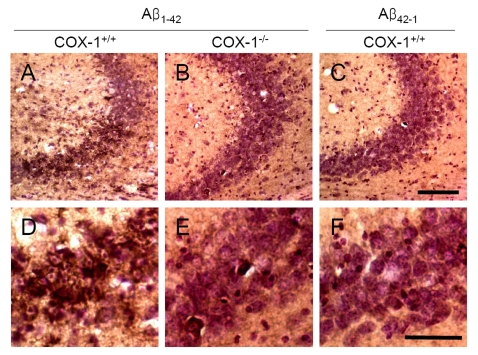
Figure 4. Increased oxidative damage in the hippocampus 7 d after Aβ 1-42 administration. Representative
photomicrographs of the CA1 and CA3 of the hippocampus from WT mice (A,
D) injected with Aβ1-42
that show numerous robustly nitrotyrosine-immunoreactive cells compared
with Aβ1-42-injected COX-1-/- mice (B,
E). Scale bar: A-C, 100 μm; D-F, 50 μm.
PG generation is reduced in Aβ1-42-injected COX-1-/- mice
To determine the contribution of COX-1 to PG production after Aβ1-42 injection, we measured the levels of PGE2, PGF2α, and TXB2 24 h after Aβ1-42 administration. We observed significant reduction in levels of PGE2 (Figure 5A), PGF2α (Figure 5B), and TXB2 (Figure 5C) inAβ1-42-injected COX-1-/- mice.
These results suggest that the reduced levels of PGE2, PGF2α, and TXB2 in COX-1-/- mice could contribute, in part, to the observed differences in glial and neuronal response to Aβ1-42 administration.
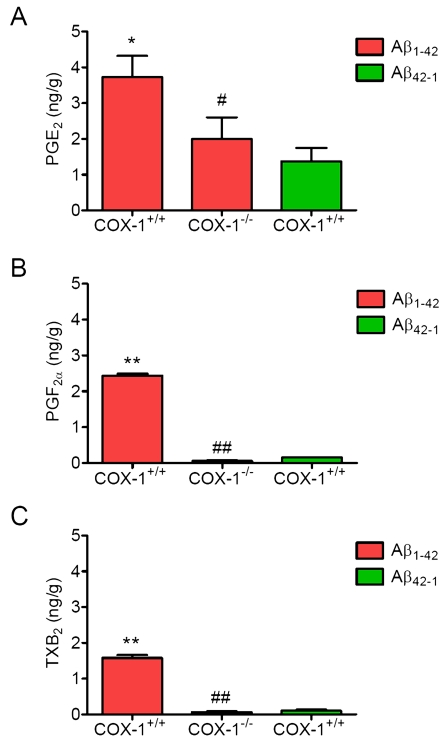
Figure 5. Effects of COX-1 deficiency on PG production 24 h after Aβ 1-42 administration. Aβ1-42-injected
WT mice show significantly more PGE2
(A), PGF2α (B), and TXB2 levels (C)
than COX-1-/- mice. Mean ± SEM (n = 3-4 per
group); *P < 0.05, **P < 0.01
compared with the Aβ42-1-injected WT mice; #P
< 0.05, ##P < 0.01 compared with the Aβ1-42-injected
WT mice.
Discussion
In this study, genetic deletion of COX-1 led to a decrease in the inflammatory response and neuronal damage in response to Aβ1-42, and this effect was associated with alteration of PG production. We show that Aβ1-42-induced oxidative damage and degenerating neurons, as well as glial activation, were less severe in COX-1-/- mice compared to WT mice. These data suggest that COX-1 facilitates activation of glial cells and supports inflammatory processes and oxidative stress that evolve in neuronal damage, and support previous data from our lab showing that COX-1-/- mice have a decreased inflammatory response, oxidative stress and neuronal damage after central injection of LPS [37].
Glial cell activation, in turn, results in enhanced production of a variety of proinflammatory and oxidative mediators, including cytokines, chemokines, and reactive oxygen/nitrogen species [40-42]. Oxidative stress has been recognized to play an important role in the pathogenesis of AD and linked to the presence of Aβ by the finding of several characteristics, such as enhanced protein, DNA oxidation, and lipid peroxidation in specific regions of the postmortem brain [43-48]. A previous study suggested that oxidative DNA damage reduces the expression of highly vulnerable genes involved in neuronal survival and learning memory, initiating a program of brain aging that starts early in adult life [49]. In addition, lipid peroxidation leads to a reduction in membrane fluidity, alteration of membrane-bounded protein, receptors, and ion channels, and generation of Aβ that induces more oxidative stress and calcium influx that induces glutamate excitotoxicity and cell death [50,51]. The abundant polyunsaturated lipid content, high oxygen consumption, high metal ion concentration, and low regenerative capacity, as well as relatively low antioxidant levels compared with other tissues make brain highly susceptible to oxidative damage [49,52]. In addition, oxidative stress differentially affects brain regions, as levels of peroxidizable unsaturated lipids and antioxidant enzymes, and membrane-bound protein differ between brain regions. In this regard, continuous icv infusion of Aβ1-42 results in a significant reduction of endogenous antioxidant systems, including Mn-superoxide dismutase (Mn-SOD), glutathione, glutathione peroxidase, and glutathione-S-transferase-π in the hippocampus, cortex, substantia nigra, and thalamus [53]. Importantly, these alterations of each antioxidant enzyme were not uniform, but rather specific in a brain region-dependent manner (e.g. Mn-SOD in CA3), indicating a heterogenous susceptibility to the Aβ1-42-induced oxidative stress.
Our results show that a single injection of Aβ1-42 resulted in a similar spatial distribution of reactive glial cells, nitrotyrosine, and degenerating neurons in the CA3 of hippocampus, suggesting the possibility that glial cell-derived reactive oxygen/nitrogen species may be involved in the impaired neuronal function, which has been described in this model [32,33,54,55]. Indeed, several studies have shown that pretreatment with antioxidants or minocycline, a tetracycline derivative with anti-inflammatory and neuroprotective properties, tend to ameliorate the Aβ1-42-induced oxidative damage and behavioral deficits [32,33,56]. Although, variable in terms of the injected Aβ peptide sequences, injection methods, and employed behavioral tests, previous studies have consistently shown the occurrence of behavioral deficits related to memory impairment after intracerebral injection of Aβ peptide [32,33,57-59]. Therefore, Aβ injection is a useful in vivo model for Aβ toxicity, which is an important component in the progression of AD.
Gene deletion of COX-1 decreased glial cell activation and attenuated nitrotyrosine induction. The decreased oxidative damage in COX-1-/- mice suggests that COX-1 deletion can reduce the activity of free-radical generating enzymes such as inducible nitric oxide (iNOS), NADPH oxidase, and myeloperoxidase (MPO). These data are consistent with recent observations that genetic deletion of COX-1 significantly reduces LPS-induced expression of both superoxide (O2-) and NO-forming enzymes and thus subsequently attenuates the levels of nitrotyrosine and protein carbonyls, which are considered as biomarkers of oxidative stress [37]. Although, the precise mechanism(s) by which COX-1 regulates free radical-generating enzymes in inflammatory cascade have not been clearly established, it is possible that because of its predominant localization in microglia, COX-1 can modulate the induction of O2-, as well as NO, from NADPH oxidase and iNOS, which, in turn, can enhance the production of more potent free radicals such as peroxynitrite (ONOO-). In addition, O2- and NO act as potent cell signaling molecules and amplify production of TNF-α and PGE2 by upregulation of COX-2 [60]. These initial effects combined with the activation of seconddary signaling cascades activate a robust immune response that consequently causes neuronal damage and death.
The results from epidemiological data indicating that NSAIDs are effective in preventing or delaying the onset of AD combined with the failure of COX-2 selective inhibitors in clinical trials in AD patients with moderate to severe AD suggest that either an early treatment is crucial to stop the mechanisms underlying the disease before the onset of the symptoms or that COX-2 selective inhibitors are not effective in delaying the progression of AD. In this regard, an intriguing hypothesis is that the protective effects of NSAIDs may be related to COX-1 rather than COX-2 inhibition. Supporting this hypothesis, COX-1 selective inhibitors (SC-560 and valeryl salicylate), but not COX-2 selective inhibitors (SC-236 and DuP-697), reduce Aβ1-42-induced PGs production and neurotoxicity in postmortem human microglia and in murine cortical neurons [61,62]. Furthermore, a small double blind, placebo-controlled study with indomethacin, a preferential COX-1 inhibitor [63], appeared to protect mild to moderately impaired AD patients from cognitive decline [14]. Interestingly, COX-1 is prominently expressed by microglia in rodent and human brain [2,4] and appears to be increased in AD brain [2]. Double immunostaining for Aβ and COX-1 indicates clustering of COX-1 positive microglia with classicaland neuritic plaques, although there is no indication that COX-1 is upregulated in activated microglia [64]. However, LPS-induced PGE2 secretion can be reduced by COX-1 genetic deletion and by COX-1 selective inhibitors [37,61,65], suggesting that it is dependent on the constitutive COX-1 activity. In contrast, COX-2 has not been detected in microglia and astrocytes in AD [66]. These combined data suggest that COX-2 may not be the exclusive COX isoform responsible for patho-physiological consequences in neurodegenerative diseases, especially in AD, but that COX-1 also plays a critical role in the process of neuroinflammation and neurodegeneration.
In summary, we show that COX-1 facilitates activation of glial cells and supports inflammatory processes and that genetic deletion of COX-1 significantly attenuates the oxidative stress and neuronal damage in response to Aβ1-42. This effect may be due to the predominant localization of COX-1 in microglial cells, where, through its prostaglandin products contributes to the neuroinflammatory cascade of events that ultimately lead to neuronal damage or death. Therefore, COX-1 may represent a viable therapeutic target to treat neuroinflammation and neurodegeneration.
Materials and methods
Animals and stereotaxic A β 1-42 administration. Three-month-old male homozygous COX-1-/- and their WT mice (COX-1+/+) on a C57BL/6-129/Ola genetic background were used [67]. Mice were received at our animal facility at 6 weeks of age from a NIEHS colony maintained by Taconic Farms (Germantown, NY) with heterozygous by heterozygous breedings for greater than 35 generations. In order to prevent the inclusion of strain or genetic background confounders between COX null and wild type mice, all of the mice used in this study were progeny derived from heterozygous by heterozygous mating and therefore all contained the same strain and genetic background [67,68]. The mice were housed at 25°C in our animal facility with a 12 h light/dark cyclewith free access to food and water. All animal procedures were approved by the National Institutes of Health (NIH) Animal Care and Use Committee in accordance with NIH guidelines on the care and use of laboratory animals. Aβ1-42 and reverse peptide Aβ42-1 (American Peptide, Sunnyvale, CA) were reconstituted in phosphate-buffered saline (pH 7.4) and aggregated by incubation at 37°C for 4 days before use as described previously [69]. Aβ1-42 and Aβ42-1 (400 pmol per mouse) were administered intracerebroventricularly (i.c.v) into the lateral ventricle using a 10 μl syringe with a fine needle (World Precision Instruments, Sarasota, FL) and a syringe pump (Stoelting, Wood Dale, IL) at a rate of 1 μl/min. The dose of Aβ1-42 and Aβ42-1 was selected based on previous studies [32-35]. The coordinates for the stereotaxic injections were -2.3mm dorsal/ventral, -1.0 mm lateral, and -0.5 mmanterior/posterior from the bregma [70].
Tissue preparation and histology. Mice were transcardially perfused withsaline followed by 4% paraformaldehyde. Brains were postfixedovernight in the same medium and placed in 30% sucrose, before sectioning (30 μm). Immunohistochemistry and double immunofluorescence were performed as described previously [71]. Rabbit anti-IBA-1 (1:500; Wako), mouse anti-GFAP (1:200; Sigma-Aldrich), and mouse anti-nitrotyrosine (1:100; Chemicon, Temecula, CA) were used as primary antibodies. The slides were visualized by brightfield microscopy (Olympus) and digitally photographed. FJB, a fluorochrome for the sensitive histochemicallocalization of neuronal degeneration, was used to identifydegenerating neurons [72]. Brainsections were mounted on gelatin-coated slides and completelydried. Then sections were rehydrated through graded concentrationsof alcohol (100, 70, and 50%; 1 min each), and rinsed for 1 min in distillated water. The slides were incubated in a solution of 0.06% potassium permanganate for 20 min, rinsedin distilled water for 1 min, and transferred to FJB (Histochem, Jefferson, AR) stainingsolution (0.001% FJB/0.1% acetic acid) for 20 min. The slides were thereafter rinsed three times in distilled water and air dried then immersedin xylene and coverslipped with mounting media. The slides were visualized by fluorescent microscopy (Olympus) and digitally photographed. Because the FJB staining was obvious on digital imaging, the number of FJB-positive cells per section was quantified as described previously [73]. The number of microglia per section was quantified by counting the number of IBA-1-stained cell bodies within 0.3 mm2 area of the CA3. For each measurement, two blinded independent investigators counted 3-4 brains per group, 3 sections per brain.
Measurement of prostanoids. Prostanoids were purified from the lipid extract as previously described [74] and levels were determined using specific enzyme immunoassay (EIA) kits, PGE2, PGF2α, and TXB2, (Oxford Biomedical, Oxford, MI).
Statistics. All data are expressed as mean ± SEM. Statistical significance was assessed with one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test using GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA). Significance was taken at P < 0.05.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health. We thank Dr. Robert Langenbach for providing COX-1-/- and WT mice. We also thank Drs. Saba Aid, Sara Palumbo, and Christopher D. Toscano for experimental suggestions and critical comments.
Conflicts of Interest
The authors in this manuscript have no conflict of interests to declare.
References
- 1. Mattson MP Pathways towards and away from Alzheimer's disease. Nature. 2004; 430: 631 -639. [PubMed] .
- 2. Yermakova AV , Rollins J , Callahan LM , Rogers J and O'Banion MK. Cyclooxygenase-1 in human Alzheimer and control brain: quantitative analysis of expression by microglia and CA3 hippocampal neurons. J Neuropathol Exp Neurol. 1999; 58: 1135 -1146. [PubMed] .
- 3. Ho L , Luterman JD , Aisen PS , Pasinetti GM , Montine TJ and Morrow JD. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology. 2000; 55: 323 [PubMed] .
- 4. Hoozemans JJ , Rozemuller AJ , Janssen I , De Groot CJ , Veerhuis R and Eikelenboom P. Cyclooxygenase expression in microglia and neurons in Alzheimer's disease and control brain. Acta Neuropathol (Berl). 2001; 101: 2 -8. [PubMed] .
- 5. Minghetti L Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004; 63: 901 -910. [PubMed] .
- 6. Combrinck M , Williams J , De Berardinis MA , Warden D , Puopolo M , Smith AD and Minghetti L. Levels of CSF prostaglandin E2, cognitive decline, and survival in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2006; 77: 85 -88. [PubMed] .
- 7. Andersen K , Launer LJ , Ott A , Hoes AW , Breteler MM and Hofman A. Do nonsteroidal anti-inflammatory drugs decrease the risk for Alzheimer's disease? The Rotterdam Study. Neurology. 1995; 45: 1441 -1445. [PubMed] .
- 8. McGeer PL , Schulzer M and McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996; 47: 425 -432. [PubMed] .
- 9. Stewart WF , Kawas C , Corrada M and Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997; 48: 626 -632. [PubMed] .
- 10. Anthony JC , Breitner JC , Zandi PP , Meyer MR , Jurasova I , Norton MC and Stone SV. Reduced prevalence of AD in users of NSAIDs and H2 receptor antagonists: the Cache County study. Neurology. 2000; 54: 2066 -2071. [PubMed] .
- 11. in t' Veld BA , Ruitenberg A , Hofman A , Launer LJ , van Duijn CM , Stijnen T , Breteler MM and Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med. 2001; 345: 1515 -1521. [PubMed] .
- 12. McGeer PL and McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. 2007; 28: 639 -647. [PubMed] .
- 13. Vlad SC , Miller DR , Kowall NW and Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008; 70: 1672 -1677. [PubMed] .
- 14. Rogers J , Kirby LC , Hempelman SR , Berry DL , McGeer PL , Kaszniak AW , Zalinski J , Cofield M , Mansukhani L and Willson P. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993; 43: 1609 -1611. [PubMed] .
- 15. Scharf S , Mander A , Ugoni A , Vajda F and Christophidis N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer's disease. Neurology. 1999; 53: 197 -201. [PubMed] .
- 16. Aisen PS , Schafer KA , Grundman M , Pfeiffer E , Sano M , Davis KL , Farlow MR , Jin S , Thomas RG and Thal LJ. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. Jama. 2003; 289: 2819 -2826. [PubMed] .
- 17. Reines SA , Block GA , Morris JC , Liu G , Nessly ML , Lines CR , Norman BA and Baranak CC. Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004; 62: 66 -71. [PubMed] .
- 18. Thal LJ , Ferris SH , Kirby L , Block GA , Lines CR , Yuen E , Assaid C , Nessly ML , Norman BA , Baranak CC and Reines SA. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005; 30: 1204 -1215. [PubMed] .
- 19. Sung S , Yang H , Uryu K , Lee EB , Zhao L , Shineman D , Trojanowski JQ , Lee VM and Pratico D. Modulation of nuclear factor-kappa B activity by indomethacin influences A beta levels but not A beta precursor protein metabolism in a model of Alzheimer's disease. Am J Pathol. 2004; 165: 2197 -2206. [PubMed] .
- 20. Kotilinek LA , Westerman MA , Wang Q , Panizzon K , Lim GP , Simonyi A , Lesne S , Falinska A , Younkin LH , Younkin SG , Rowan M , Cleary J , Wallis RA , Sun GY , Cole G , Frautschy S , Anwyl R and Ashe KH. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. 2008; 131: 651 -664. [PubMed] .
- 21. Jantzen PT , Connor KE , DiCarlo G , Wenk GL , Wallace JL , Rojiani AM , Coppola D , Morgan D and Gordon MN. Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002; 22: 2246 -2254. [PubMed] .
- 22. Kukar T , Murphy MP , Eriksen JL , Sagi SA , Weggen S , Smith TE , Ladd T , Khan MA , Kache R , Beard J , Dodson M , Merit S , Ozols VV , Anastasiadis PZ , Das P , Fauq A , Koo EH and Golde TE. Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med. 2005; 11: 545 -550. [PubMed] .
- 23. Aid S , Langenbach R and Bosetti F. Neuroinflammatory response to lipopolysaccharide is exacerbated in mice genetically deficient in cyclooxygenase-2. J Neuroinflammation. 2008; 5: 17 [PubMed] .
- 24. Eriksen JL , Sagi SA , Smith TE , Weggen S , Das P , McLendon DC , Ozols VV , Jessing KW , Zavitz KH , Koo EH and Golde TE. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003; 112: 440 -449. [PubMed] .
- 25. Gasparini L , Rusconi L , Xu H , del Soldato P and Ongini E. Modulation of beta-amyloid metabolism by non-steroidal anti-inflammatory drugs in neuronal cell cultures. J Neurochem. 2004; 88: 337 -348. [PubMed] .
- 26. Morihara T , Teter B , Yang F , Lim GP , Boudinot S , Boudinot FD , Frautschy SA and Cole GM. Ibuprofen suppresses interleukin-1beta induction of pro-amyloidogenic alpha1-antichymotrypsin to ameliorate beta-amyloid (Abeta) pathology in Alzheimer's models. Neuropsychopharmacology. 2005; 30: 1111 -1120. [PubMed] .
- 27. Peretto I , Radaelli S , Parini C , Zandi M , Raveglia LF , Dondio G , Fontanella L , Misiano P , Bigogno C , Rizzi A , Riccardi B , Biscaioli M , Marchetti S , Puccini P , Catinella S , Rondelli I , Cenacchi V , Bolzoni PT , Caruso P , Villetti G , Facchinetti F , Del Giudice E , Moretto N and Imbimbo BP. Synthesis and biological activity of flurbiprofen analogues as selective inhibitors of beta-amyloid(1)(-)(42) secretion. J Med Chem. 2005; 48: 5705 -5720. [PubMed] .
- 28. Kukar TL , Ladd TB , Bann MA , Fraering PC , Narlawar R , Maharvi GM , Healy B , Chapman R , Welzel AT , Price RW , Moore B , Rangachari V , Cusack B , Eriksen J , Jansen-West K , Verbeeck C , Yager D , Eckman C , Ye W , Sagi S , Cottrell BA , Torpey J , Rosenberry TL , Fauq A , Wolfe MS , Schmidt B , Walsh DM , Koo EH and Golde TE. Substrate-targeting gamma-secretase modulators. Nature. 2008; 453: 925 -929. [PubMed] .
- 29. Szekely CA , Green RC , Breitner JC , Ostbye T , Beiser AS , Corrada MM , Dodge HH , Ganguli M , Kawas CH , Kuller LH , Psaty BM , Resnick SM , Wolf PA , Zonderman AB , Welsh-Bohmer KA and Zandi PP. No advantage of A beta 42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology. 2008; 70: 2291 -2298. [PubMed] .
- 30. Yamada K and Nabeshima T. Animal models of Alzheimer's disease and evaluation of anti-dementia drugs. Pharmacol Ther. 2000; 88: 93 -113. [PubMed] .
- 31. Bosetti F Arachidonic acid metabolism in brain physiology and pathology: lessons from genetically altered mouse models. J Neurochem. 2007; 102: 577 -586. [PubMed] .
- 32. Yan JJ , Cho JY , Kim HS , Kim KL , Jung JS , Huh SO , Suh HW , Kim YH and Song DK. Protection against beta-amyloid peptide toxicity in vivo with long-term administration of ferulic acid. Br J Pharmacol. 2001; 133: 89 -96. [PubMed] .
- 33. Jhoo JH , Kim HC , Nabeshima T , Yamada K , Shin EJ , Jhoo WK , Kim W , Kang KS , Jo SA and Woo JI. Beta-amyloid (1-42)-induced learning and memory deficits in mice: involvement of oxidative burdens in the hippocampus and cerebral cortex. Behav Brain Res. 2004; 155: 185 -196. [PubMed] .
- 34. Prediger RD , Franco JL , Pandolfo P , Medeiros R , Duarte FS , Di Giunta G , Figueiredo CP , Farina M , Calixto JB , Takahashi RN and Dafre AL. Differential susceptibility following beta-amyloid peptide-(1-40) administration in C57BL/6 and Swiss albino mice: Evidence for a dissociation between cognitive deficits and the glutathione system response. Behav Brain Res. 2007; 177: 205 -213. [PubMed] .
- 35. Medeiros R , Prediger RD , Passos GF , Pandolfo P , Duarte FS , Franco JL , Dafre AL , Di Giunta G , Figueiredo CP , Takahashi RN , Campos MM and Calixto JB. Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer's disease: relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J Neurosci. 2007; 27: 5394 -5404. [PubMed] .
- 36. Schmued LC , Albertson C and Slikker W Jr. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997; 751: 37 -46. [PubMed] .
- 37. Choi SH , Langenbach R and Bosetti F. Genetic deletion or pharmacological inhibition of cyclooxygenase-1 attenuate lipopolysaccharide-induced inflammatory response and brain injury. Faseb J. 2008; 22: 1491 -1501. [PubMed] .
- 38. Yan SD , Chen X , Fu J , Chen M , Zhu H , Roher A , Slattery T , Zhao L , Nagashima M , Morser J , Migheli A , Nawroth P , Stern D and Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996; 382: 685 -691. [PubMed] .
- 39. Liberatore GT , Jackson-Lewis V , Vukosavic S , Mandir AS , Vila M , McAuliffe WG , Dawson VL , Dawson TM and Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999; 5: 1403 -1409. [PubMed] .
- 40. Block ML , Zecca L and Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007; 8: 57 -69. [PubMed] .
- 41. McGeer PL , Itagaki S , Tago H and McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987; 79: 195 -200. [PubMed] .
- 42. Nelson PT , Soma LA and Lavi E. Microglia in diseases of the central nervous system. Ann Med. 2002; 34: 491 -500. [PubMed] .
- 43. Butterfield DA and Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002; 32: 1050 -1060. [PubMed] .
- 44. Castegna A , Thongboonkerd V , Klein JB , Lynn B , Markesbery WR and Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003; 85: 1394 -1401. [PubMed] .
- 45. Hensley K , Maidt ML , Yu Z , Sang H , Markesbery WR and Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998; 18: 8126 -8132. [PubMed] .
- 46. Markesbery WR and Carney JM. Oxidative alterations in Alzheimer's disease. Brain Pathol. 1999; 9: 133 -146. [PubMed] .
- 47. Smith MA , Richey Harris PL , Sayre LM , Beckman JS and Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997; 17: 2653 -2657. [PubMed] .
- 48. Sultana R , Perluigi M and Butterfield DA. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer's disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal. 2006; 8: 2021 -2037. [PubMed] .
- 49. Lu T , Pan Y , Kao SY , Li C , Kohane I , Chan J and Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004; 429: 883 -891. [PubMed] .
- 50. Lauritzen L , Hansen HS , Jorgensen MH and Michaelsen KF. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog Lipid Res. 2001; 40: 1 -94. [PubMed] .
- 51. Yehuda S , Rabinovitz S and Mostofsky DI. Essential fatty acids are mediators of brain biochemistry and cognitive functions. J Neurosci Res. 1999; 56: 565 -570. [PubMed] .
- 52. Lane RM and Farlow MR. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer's disease. J Lipid Res. 2005; 46: 949 -968. [PubMed] .
- 53. Kim HC , Yamada K , Nitta A , Olariu A , Tran MH , Mizuno M , Nakajima A , Nagai T , Kamei H , Jhoo WK , Im DH , Shin EJ , Hjelle OP , Ottersen OP , Park SC , Kato K , Mirault ME and Nabeshima T. Immunocytochemical evidence that amyloid beta (1-42) impairs endogenous antioxidant systems in vivo. Neuroscience. 2003; 119: 399 -419. [PubMed] .
- 54. Weldon DT , Rogers SD , Ghilardi JR , Finke MP , Cleary JP , O'Hare E , Esler WP , Maggio JE and Mantyh PW. Fibrillar beta-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci. 1998; 18: 2161 -2173. [PubMed] .
- 55. Klein AM , Kowall NW and Ferrante RJ. Neurotoxicity and oxidative damage of beta amyloid 1-42 versus beta amyloid 1-40 in the mouse cerebral cortex. Ann N Y Acad Sci. 1999; 893: 314 -320. [PubMed] .
- 56. Ryu JK and McLarnon JG. Minocycline or iNOS inhibition block 3-nitrotyrosine increases and blood-brain barrier leakiness in amyloid beta-peptide-injected rat hippocampus. Exp Neurol. 2006; 198: 552 -557. [PubMed] .
- 57. O'Hare E , Weldon DT , Mantyh PW , Ghilardi JR , Finke MP , Kuskowski MA , Maggio JE , Shephard RA and Cleary J. Delayed behavioral effects following intrahippocampal injection of aggregated A beta (1-42). Brain Res. 1999; 815: 1 -10. [PubMed] .
- 58. Nakamura S , Murayama N , Noshita T , Annoura H and Ohno T. Progressive brain dysfunction following intracerebroventricular infusion of beta(1-42)-amyloid peptide. Brain Res. 2001; 912: 128 -136. [PubMed] .
- 59. Christensen R , Marcussen AB , Wortwein G , Knudsen GM and Aznar S. Abeta(1-42) injection causes memory impairment, lowered cortical and serum BDNF levels, and decreased hippocampal 5-HT(2A) levels. Exp Neurol. 2008; 210: 164 -171. [PubMed] .
- 60. Qin L , Liu Y , Wang T , Wei SJ , Block ML , Wilson B , Liu B and Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004; 279: 1415 -1421. [PubMed] .
- 61. Hoozemans JJ , Veerhuis R , Janssen I , van Elk EJ , Rozemuller AJ and Eikelenboom P. The role of cyclo-oxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: implications for Alzheimer's disease. Brain Res. 2002; 951: 218 -226. [PubMed] .
- 62. Bate C , Veerhuis R , Eikelenboom P and Williams A. Neurones treated with cyclo-oxygenase-1 inhibitors are resistant to amyloid-beta1-42. Neuroreport. 2003; 14: 2099 -2103. [PubMed] .
- 63. Barnett J , Chow J , Ives D , Chiou M , Mackenzie R , Osen E , Nguyen B , Tsing S , Bach C and Freire J. Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochim Biophys Acta. 1994; 1209: 130 -139. [PubMed] .
- 64. Hoozemans JJ , Rozemuller JM , van Haastert ES , Veerhuis R and Eikelenboom P. Cyclooxygenase-1 and -2 in the different stages of Alzheimer's disease pathology. Curr Pharm Des. 2008; 14: 1419 -1427. [PubMed] .
- 65. Candelario-Jalil E , Taheri S , Yang Y , Sood R , Grossetete M , Estrada EY , Fiebich BL and Rosenberg GA. Cyclooxygenase inhibition limits blood-brain barrier disruption following intracerebral injection of tumor necrosis factor-alpha in the rat. J Pharmacol Exp Ther. 2007; 323: 488 -498. [PubMed] .
- 66. Hoozemans JJ , Veerhuis R , Rozemuller AJ and Eikelenboom P. Non-steroidal anti-inflammatory drugs and cyclooxygenase in Alzheimer's disease. Curr Drug Targets. 2003; 4: 461 -468. [PubMed] .
- 67. Langenbach R , Morham SG , Tiano HF , Loftin CD , Ghanayem BI , Chulada PC , Mahler JF , Lee CA , Goulding EH , Kluckman KD , Kim HS and Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995; 83: 483 -492. [PubMed] .
- 68. Toscano CD , Prabhu VV , Langenbach R , Becker KG and Bosetti F. Differential gene expression patterns in cyclooxygenase-1 and cyclooxygenase-2 deficient mouse brain. Genome Biol. 2007; 8: R14 [PubMed] .
- 69. Maurice T , Lockhart BP and Privat A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996; 706: 181 -193. [PubMed] .
- 70. Paxinos G and Franklin KBJ. San Diego, Calif, London Academic The mouse brain in stereotaxic coordinates. edn 2nd. 2001; .
- 71. Choi SH , Lee da Y , Kim SU and Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci. 2005; 25: 4082 -4090. [PubMed] .
- 72. Schmued LC and Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000; 874: 123 -130. [PubMed] .
- 73. El Khoury J , Toft M , Hickman SE , Means TK , Terada K , Geula C and Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007; 13: 432 -438. [PubMed] .
- 74. Powell WS Reversed-phase high-pressure liquid chromatography of arachidonic acid metabolites formed by cyclooxygenase and lipoxygenases. Anal Biochem. 1985; 148: 59 -69. [PubMed] .