



miRNAs are key regulators of cell fate
miRNAs have emerged in the past decade as important players in numerous cellular and organismal processes in animals and plants [1]. Deletion of the Dicer gene, encoding the critical enzyme involved in miRNA processing and maturation, is embryonic lethal in both mice [2] and zebrafish [3]. Accordingly, many studies showed, using conditional elimination of Dicer, that miRNAs are crucial for the proper spatiotemporal development of various tissues and organs ([2,4-9] and reviewed in [10]). Further, mouse embryonic stem (ES) cells defective in miRNA processing were shown to proliferate slower [11], and to be impaired in their ability to differentiate [8]. In parallel, other studies have shown a major role for miRNAs in development, indicating that many miRNAs are upregulated during the process of ES cell differentiation ([12] and reviewed in [13]). Many miRNAs also play a role in differentiation processes in the adult organism, including hematopoiesis [14] and the germinal center response [15]. In fact, the first miRNAs to be discovered, lin-4 and let-7 in C. elegans, regulate epithelial cell differentiation [16,17]. In addition, manipulations of individual miRNA genes were shown to result in marked defects at the organismal level ([18,19] and reviewed in [20]). Based on these accumulated observations it is plausible to suggest that in many cases miRNAs are indeed a part of the driving force of differentiation processes. miRNAs were also shown to regulate many cellular processes [21,22], such as cell growth and proliferation (reviewed in [23,24]) and apoptosis (reviewed in [25]). It appears, therefore, that miRNAs are crucial players in the regulation and determination of cell fate.
miRNAs - guardians of genome integrity?
Lu et al. [26] carried out an extensive analysis of miRNA expression in human cancer. This study, that included a global expression profiling of miRNAs across a large set of tumors, demonstrated that miRNA expression profiles can be used to classify human cancers of unknown origin. In addition, the researchers made the very interesting observation that, in general, tumors have lower levels of miRNAs than normal tissues. The authors suggested that the observed low global levels of miRNAs may be a reflection of the de-differentiated state of tumors.
An alternative, complementary explanation might be that tumors evolve to silence the miRNA pathway during the course of cancer progression. In other words, globally avoiding regulation of gene expression by miRNAs may be one of the many ways of cancer cells to enhance their proliferation and tumorigenic potential.
Several lines of evidence support the idea that proliferating cells and cancer cells in particular, find many different ways to avoid post-transcriptional regulation by miRNAs (Figure 1). Some of these mechanisms are straightforward, and are in agreement with what we know of tumor suppressors and oncogenes. For example, the MYC oncogenic transcription factor (TF) was found in a lymphoma mouse model to mediate widespread repression of a large set of miRNAs, contributing to tumorigenesis [27]. Other mechanistic possibilities for tumors to avoid posttranscriptional regulation by miRNAs include epigenetic silencing, mutation and deletion of genomic loci encoding for miRNAs [28-33]. A prominent example is the miR-15a/16-1 cluster, residing in the DLEU2 non-coding RNA, which was long known to be frequently deleted in leukemia [34,35], and was later shown to harbor these miRNAs [29]. Another newly described mechanism is the interruption of the miRNA biogenesis pathway, by processes such as nuclear retention of unprocessed pre-miRNAs [36], or pri- and pre-miRNA processing blockage such as in the case of inhibition of maturation of the let-7 family by the Lin28 protein [37-39]. Lin28 was further shown to promote cancer, and this was attributed to its repression of the let-7 miRNA family [40]. A recent report implicates p53 in the enhancement of miRNA maturation for many miRNAs following DNA damage [41], attesting to global miRNA upregulation as a possible anti-cancer mechanism. Additional highly intriguing phenomenon was reported by Sandberg et al. [42], indicating that proliferating cells tend to employ alternative polyadenylation or alternative splicing in order to express mRNAs with shorter 3' UTRs, having fewer miRNA binding sites. These shorter mRNAs avoid post-transcriptional regulation by miRNAs, thus potentially enhancing their protein level. This phenomenon represents another path by which proliferating cells achieve the same goal - avoiding miRNA-mediated silencing, presumably in order to accelerate proliferation.
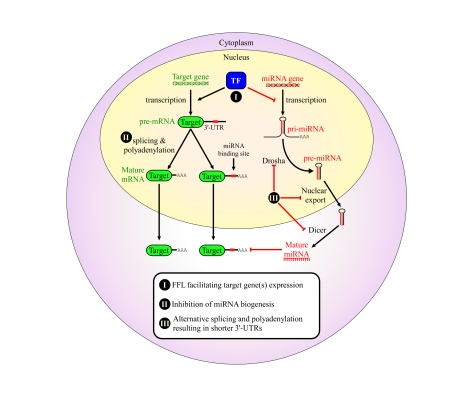
Figure 1. Proposed mechanisms for avoidance of regulation by miRNAs in cancer cells.
We propose that cancers may
evolve to avoid regulation by miRNAs in order to enhance their tumorigenic
potential. This might occur through a variety of mechanisms: (I) combined
transcriptional/post-transcriptional FFL wiring, which may enhance the
repression of several co-regulated miRNAs, thereby facilitating the
expression of the mutual target genes; (II) global avoidance of miRNA
regulation via expression of shorter 3' UTRs [42]; (III) global reduction
in miRNA levels by impairing miRNA biogenesis in various ways, some of
which were shown to happen in tumors, such as inhibition of Drosha processing [39,40] and pre-miRNA nuclear retention [36]. All of these are suggested as
means that developing tumors may evolve to enhance proliferation and
increase genome instability.
The most striking evidence in support of the 'miRNA avoidance' strategy played by tumors is shown by two seemingly contradictory studies, one focusing on cancer cells and the other on normal cells. The study by Kumar et al. [43] reported that the ablation of miRNAs in various cancer cell lines resulted in enhanced cellular transformation, evident by increased colony formation efficiency in vitro and increased tumor burden in vivo. On the other hand, Mudhasani et al. [44] showed that the total elimination of miRNAs using conditional Dicer knock-out results in premature senescence in normal mouse embryonic fibroblasts (MEFs). This effect was also apparent at the level of the organism, as the knock-out ofDicer in keratinocytes and skin epidermis of adult mice resulted in senescence-induced hairloss and skin aging [44].
At first glance, these two studies seem to disagree. How is it possible that a similar manipulation would enhance proliferation in one system, and cause a proliferation arrest or senescence in the other? A potential solution to this conflict would consider that the same event can lead to two opposite outcomes, depending on the cellular context. For example, activation of an oncogene, such as RAS, is one of the hallmarks of cancer, and when occurring in cancer cells will cause the enhancement of their tumorigenic phenotype. However, in normal cells, oncogene activation will often lead to genomic instability, which is sensed by the DNA damage checkpoint, and leads to p53 and ARF-dependent senescence, a phenomenon known as "oncogene-induced senescence" [45]. Importantly, the phenomenon described by Mudhasani et al. [44] was not a classical case of oncogene-induced senescence, as it was not accompanied by the upregulation of the oncogenes MYC or RAS, (two well known activators of oncogene-induced senescence), even though they are documented miRNA targets [46-48]. Interestingly, however, the depletion of miRNAs led to DNA damage, as evident by γH2A.X staining, and consequently, through activation of the p19ARF and p53-dependent DNA-damage checkpoint, resulted in premature senescence.
Therefore, in this case too, the same event of global miRNA depletion induced the DNA damage checkpoint in normal cells due to proper p19ARF and p53 activation, while in cancer cells it led to enhanced transformation, where these checkpoint response pathways are frequently inactivated, and genomic instability enhances tumorigenesis [49].
Importantly, as we outline here, inactivation of miRNA-mediated silencing is not only capable in principle of influencing cell fate, following genetic manipulations as shown by Mudhasani et al. and Kumar et al. [43,44], but may actually occur in vivo during tumorigenesis [26,42]. It therefore seems likely that miRNAs are not only necessary for proliferation and differentiation in normal cells, but also act to maintain normal cell proliferation, and may be thought of as "guardians" of genome integrity. In cancer cells, on the other hand, inactivation of the miRNA-mediated silencing pathway and the avoidance of miRNA regulation contribute to transformation (Figure 1). In principle we can therefore consider miRNAs as a regulatory barrier whose removal may be part of a series of events that ultimately lead to cancer.
A conceptual gap between the influence of miRNAs on protein levels and their effects on cell fate
miRNAs can exert their silencing effects by cleavage of their target mRNAs and by inhibition of their translation. A common knowledge in the field was that animal miRNAs exert most of their silencing through the inhibition of translation, rather than through the degradation of their targets, and that this was due to a low overall degree of sequence complementarity that animal miRNAs share with their target sites on 3' UTRs of mRNAs [1]. In fact, the first discovered miRNAs in C. elegans, lin-4, was shown to inhibit the translation of its target Lin-14, without affecting its mRNA levels [50,51]. Mechanistically, it became evident that the miRNA-effector protein complex, the RISC, is enzymatically capable of both mRNA cleavage and inhibition of translation [52,53]. Lim et al. then showed that miRNAs can influence the mRNA levels of their target genes [54]. Using overexpression of miRNAs followed by global expression profiling using microarrays, they demonstrated a modest but significant downregulation of mRNA levels of genes that were enriched for the miRNA seed sequence. This study and others that followed contributed to the overall view that miRNAs exert silencing through both mechanisms simultaneously, but the more major effect was expected at the protein level, rather than at the mRNA levels.
Recent studies used high throughput proteomics in order to both identify translationally inhibited targets and to more accurately assess the extent of inhibition that a miRNA exerts on mRNA levels and on protein levels [55,56]. These studies reported that individual miRNAs affect hundreds of proteins in the human and mouse out of thousands that were examined. However, the levels of these proteins were decreased only to a relatively mild extent. miRNAs were often before considered as modulators of expression, and their generally observed mild effect on protein levels (and mRNA levels as well) promoted their suggested role as buffers for noise in protein expression, which may confer robustness to developmental programs [57].
Overall, there seems to be a discrepancy between the observation that miRNAs have such subtle effects on protein levels and the fact that their effects on cell fate are so profound. We would like to suggest here one possible model that might bridge this conceptual gap.
Coupling transcriptional and post-transcriptional miRNA regulation in the control of cell fate
One trivial way to resolve the above discrepancy might argue that the multiplicity of miRNA targets and the simultaneous down-regulation of many proteins might have a cumulative effect, eventually exerting a significant impact on cell fate, even though individual proteins are repressed to a very modest extent. This is a valid argument, particularly since some miRNAs were predicted and shown to have multiple targets within the same pathway [58-60], thus potentially having greater effects on entire pathways than on individual proteins.
While miRNAs may exert modest effects, yet on many targets, another possible answer to their significant effect on cell fate may lie in the level of the regulatory networks that miRNAs take central part in. miRNAs do not act in isolation, but rather they regulate target genes combinatorially with one another, and are often embedded within intricate regulatory networks together with TFs (Figure 2). In fact, it was demonstrated that at the network level, there is tight coupling between posttranscriptional regulation by miRNAs and the regulation of transcription by TFs [61,62]. Examination of regulatory networks showed that in many cases the same TF controls the transcription of both a miRNA and the targets of that miRNA, or is regulated by the same miRNA with which it shares common targets, forming a diversity of combined transcriptional/post-transcriptional Feed-Forward Loops (FFLs). Collectively, such FFLs potentially regulate thousands of target genes.
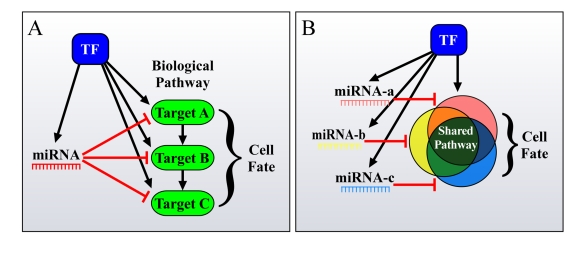
Figure 2. Different ways by which FFLs can account for the enhanced phenotypic effect of miRNAs on cell fate. (A) miRNAs
and TFs in FFLs tend to mutually target genes from the same pathway. (B)
Additionally, co-regulated miRNAs and miRNA families co-target many genes
in the same pathway, thus resulting in a significant total output, having a
major effect on cell fate.
Network analyses showed that these FFLs constitute over-represented architectures in the mammalian regulatory network [61,62]. Network FFLs, initially described by Alon and colleagues, were shown to comprise a major component of the transcription networks in bacteria and yeast [63,64]. The discovery that miRNAs and TFs also constitute FFLs offered new possibilities for potential functions for these regulatory units. Clues for the existence of coupling between transcription and miRNA regulation emerged from a very intriguing concept, called miRNA-target avoidance. Two parallel studies, one in Drosophila and the other in mammals, showed that during development as well as in adult tissues, miRNA targets often avoid being expressed in the same tissue, or at the same developmental time, as their potential inhibitory miRNA [65,66]. In Drosophila, it was shown for some cases that a miRNA and its targets are expressed in adjacent tissues during development, or in consecutive developmental stages, and that miRNAs serve as key players in the precise definition of spatiotemporal differentiation boundaries [66]. This phenomenon was observed also in adult tissues and organs in both Drosophila [66] and mouse [65]. Moreover, both studies indicated that this mutual exclusion of miRNAs and their targets does not stem from target degradation by the miRNA. From these two studies, it became evident that posttranscriptional regulation by miRNAs is somehow coordinated with transcription. However, it was not shown originally how, at the mechanistic level, such "miRNA-target spatiotemporal avoidance" is achieved. Combined transcriptional/posttranscriptional FFLs, where the same TF regulates the transcription of both a miRNA and its target genes, or where the miRNA targets a TF and its target genes as well, could serve just that purpose (Figure 3). Such FFLs are thus suggested as a simple mechanism that might facilitate the miRNA-target avoidance phenomenon, where a TF that activates the target genes also represses the miRNA transcription in the tissues in which it is expressed, or the miRNA represses both the TF and its target genes, thereby indirectly causing reduced transcription of its targets in the tissue where it is expressed (Figure 3) [61]. In addition, such FFLs were further suggested to enable the "canalization" and the maintenance of fidelity of developmental processes in general [57].
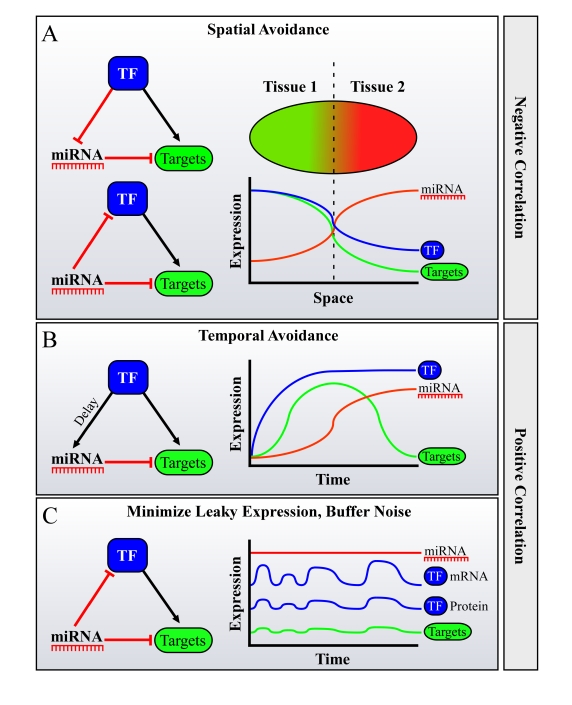
Figure 3. Possible roles for FFLs of miRNAs, Transcription Factors (TFs) and their mutual targets in facilitating spatiotemporal avoidance, or noise buffering. miRNAs are often
embedded in Feed-Forward loops (FFLs) with TFs, sharing mutual targets. It
was shown that in many cases during development, miRNAs and their targets
avoid expression in the same tissue or at the same developmental stage.
This phenome-non was termed "miRNA-target spatiotemporal
avoidance". The figure depicts how the network wiring of miRNAs in
combined transcriptional/posttranscriptional FFLs may explain the spatio-temporal
avoidance phenomenon. Different scenarios may facilitate spatial and
temporal avoidance, where the TF and the miRNA are either negatively
correlated in their expression across tissues (in A) or positively
correlated, namely are expressed in the same tissue (B or C).
(A) Spatial
avoidance may be facilitated by the presented FFLs when expression of a
miRNA and of a TF anti-correlates across tissues. (B) Temporal avoidance
may be facilitated by the presented FFL when a miRNA and a TF are
co-expressed in the same tissues, creating a temporal shut-down mechanism
for their mutual targets, when there is a delay between the activation of
the targets by the TF, and its activation of the miRNA. This delay may be
achieved for example by a lower affinity binding site of the TF to the
miRNA's promoter, by a natural miRNA processing time, etc. (C) Buffering of
noise in expression may also be facilitated by a FFL wiring when a miRNA and
a TF are co-expressed in the same tissues.
More recently, evidence has been accumulating that such combined transcriptional post-transcriptional FFLs indeed act as functional units in the regulation of cell fate in many cell types and systems [48,58,67-71]. One striking example, recently published by Marson et al. [69], demonstrated that miRNAs and TFs are involved together in FFLs controlling the maintenance of mouse embryonic stem (ES) cell identity. Consistent with the studies mentioned above [2,3,8,11], which showed that complete miRNA ablation from ES cells eliminates their differentiation capacity, Marson et al. showed that several FFLs involving miRNAs and ES cell TFs act to regulate ES cell identity and differentiation. For example, the miR-290-295 polycistronic cluster, containing the most abundantly expressed miRNAs in mouse ES cells, is positively regulated by the ES cell TF Oct4, whereas its promoter is co-occupied by Oct4, Sox2, and Nanog. In addition, miR-290-295 co-regulate mutual target genes along with these same TFs. Intriguingly, while miR-290-295 is a rodent specific cluster, a similar FFL involving Sox and Oct4 was computationally predicted in humans [61]. This FFL comprises miR-302, which shares the same seed as the rodent-specific miR-290-295, and was shown to be highly expressed in human ES cells [72], perhaps serving as a miR-290-295 human ortholog. Consideration of these results in the perspective of previous studies on miRNAs role in ES cell differentiation supports the conjecture that miRNA-involving FFLs might play an important role in this context, and suggest potential conserved roles for similar FFLs in the maintenance of human ES cell identity as well.
A different perspective on miRNA-TF FFLs was recently provided by Brosh et al. [58]. In this study, a family of 15 homologous miRNAs transcribed as three polycistrons: miR-106b/93/-25, miR-17-92 and miR-106a-363, were shown to form a proliferation-promoting FFL together with the transcription factor E2F. These miRNAs were shown to target a whole battery of anti-proliferative E2F target genes. Most importantly, the study demonstrated that in normal fibroblasts p53 inhibits this FFL as a central step towards cellular senescence. When this inhibition is perturbed by overexpression of the miRNAs, normal cell fate is altered; proliferation is accelerated and senescence is delayed. In agreement with these results, breast cancer tumors bearing mutated p53 showed an elevation in the levels of these miRNAs and were characterized by a high tumor grade, hinting at the role of these miRNAs in promoting proliferation and aggressiveness also in vivo in tumors. This miRNA family was indeed reported in several independent studies to be related to promotion of cancer [58,73,74] (also reviewed in [75]). The above study illustrates how deregulation of the entire FFL may contribute to aberrant proliferation. It also reveals another concept of network wiring of miRNAs, namely combinatorial regulation, and more specifically combinatorial regulation by family-related miRNAs (Figure 2). Combinatorial regulation by miRNAs was globally predicted based on co-occurrence of miRNA target sites in common gene sets [61], and was also observed experimentally [58,76].
miRNAs can be grouped by mature sequence similarity into miRNA families. In some cases, as in the case of the miR-106b/93/-25 family mentioned above, these families are shown to represent paralogous groups of miRNAs of a common evolutionary origin [77]. Just as paralogous genes were duplicated during evolution but retained some degree of sequence similarity, these paralogous miRNAs share similarity in their sequence, which immediately suggests that they might also share common target genes. More intriguingly, it seems that in many cases such families had not only retained similar targets, but also retained similar transcriptional programs. As described by Brosh et al. [58], the above family of 15 miRNAs retained their joint transcriptional regulation by E2F. Coordinated transcriptional regulation of a family of miRNAs, sharing similar targets, all of which are part of the same pathway (in this case negative regulators of proliferation), may have a cumulative effect on the overall levels of proteins in the pathway, thus resulting in a strong effect on cell fate.
Coordinated regulation of family miRNAs was also shown in other cases [78,79]. For example the miR-34 family, consisting of two transcription units and three mature family members, were all shown to be transcriptionally activated by p53 and to contribute to apoptosis [80,81], G1 cell-cycle arrest [82] and senescence [83]. Moreover, miR-34a and miR-34c were shown to target c-MYC [46, 84]. In addition, in both mouse and human ES cells, several related miRNA families, often sharing similar seeds, were shown to be co-expressed [69,72]. Moreover, miRNAs from the same family were indeed verified experimentally to have many shared targets [76].
Overall it seems that combinatorial regulation of miRNAs, particularly from the same family, and shared transcription programs for such miRNAs and their common targets portray intricate network architecture (Figure 2). Such architecture is not only over-represented [61], but may also cumulatively generate a strong output that is likely to account for the observed effects on cell fate, and for its alteration when the miRNAs are mis-regulated.
Concluding Remarks
It is intriguing that despite a relatively mild influence of individual miRNAs on protein levels they are indispensable to various cellular and organismal processes, including control of cell fate and maintenance of genomic integrity. One possible explanation for this may lie in the level of regulatory networks in which miRNAs are embedded. Indeed, joint miRNA-TF FFLs are not only an over-represented architecture in the network but a recurring principle of miRNA regulation of cell fate.
The connection between cell fate and the wiring of miRNAs in coupled transcription/post-transcriptional networks is appealing, and the multiple evidence outlines here serve to support it.
Two principles are common to the different examples discussed above: 1. miRNAs are embedded in combined transcription-nal/post-transcriptional FFLs that co-target many genes.
2. Several co-regulated miRNAs act together to exert their regulation on target genes involved in the same pathway.
However, more studies should be undertaken in order to fully establish the link between the network wiring of miRNAs in transcriptional/post-transcriptional FFLs and their effect on cell fate. A recent study demonstrated that the wiring of miR-7 in a network of FFLs in the fly equips the network with robustness to environmental perturbation [68]. Such approach suggests that when studying possible roles for miRNAs, one should consider them as parts of a larger regulatory network, rather than adopting the reductionist view of single miRNA - single target. Our recognition of the centrality of miRNAs in the regulatory network may help us to elucidate how miRNAs exert such profound impact on cell fate.
Conflicts of Interest
The authors declare no conflict of interests.
References
- 1. Bartel DP MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116: 281 -297. [PubMed] .
- 2. Bernstein E , Kim SY , Carmell MA , Murchison EP , Alcorn H , Li MZ , Mills AA , Elledge SJ , Anderson KV and Hannon GJ. Dicer is essential for mouse development. Nat Genet. 2003; 35: 215 -217. [PubMed] .
- 3. Wienholds E , Koudijs MJ , van Eeden FJ , Cuppen E and Plasterk RH. The microRNA-producing enzyme Dicer1 is essential for zebrafish development. Nat Genet. 2003; 35: 217 -218. [PubMed] .
- 4. Chen JF , Murchison EP , Tang R , Callis TE , Tatsuguchi M , Deng Z , Rojas M , Hammond SM , Schneider MD , Selzman CH , Meissner G , Patterson C and Hannon GJ. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2008; 105: 2111 -2116. [PubMed] .
- 5. Damiani D , Alexander JJ , O'Rourke JR , McManus M , Jadhav AP , Cepko CL , Hauswirth WW , Harfe BD and Strettoi E. Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J Neurosci. 2008; 28: 4878 -4887. [PubMed] .
- 6. Davis TH , Cuellar TL , Koch SM , Barker AJ , Harfe BD , McManus MT and Ullian EM. Conditional loss of Dicer disruptscellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci. 2008; 28: 4322 -4330. [PubMed] .
- 7. Volinia S , Calin GA , Liu CG , Ambs S , Cimmino A , Petrocca F , Visone R , Iorio M , Roldo C , Ferracin M , Prueitt RL , Yanaihara N and Lanza G. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006; 103: 2257 -2261. [PubMed] .
- 8. Kanellopoulou C , Muljo SA , Kung AL , Ganesan S , Drapkin R , Jenuwein T , Livingston DM and Rajewsky K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005; 19: 489 -501. [PubMed] .
- 9. Murchison EP , Stein P , Xuan Z , Pan H , Zhang MQ , Schultz RM and Hannon GJ. Critical roles for Dicer in the female germline. Genes Dev. 2007; 21: 682 -693. [PubMed] .
- 10. Bushati N and Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007; 23: 175 -205. [PubMed] .
- 11. Murchison EP , Partridge JF , Tam OH , Cheloufi S and Hannon GJ. Characterization of Dicer-deficient murine embryonic stem cells. Proc Natl Acad Sci U S A. 2005; 102: 12135 -12140. [PubMed] .
- 12. Houbaviy HB , Murray MF and Sharp PA. Embryonic stem cell-specific MicroRNAs. Dev Cell. 2003; 5: 351 -358. [PubMed] .
- 13. Gangaraju VK and Lin H. MicroRNAs: key regulators of stem cells. Nat Rev Mol Cell Biol. 2009; 10: 116 -125. [PubMed] .
- 14. Lu J , Guo S , Ebert BL , Zhang H , Peng X , Bosco J , Pretz J , Schlanger R , Wang JY , Mak RH , Dombkowski DM , Preffer FI and Scadden DT. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev Cell. 2008; 14: 843 -853. [PubMed] .
- 15. Thai TH , Calado DP , Casola S , Ansel KM , Xiao C , Xue Y , Murphy A , Frendewey D , Valenzuela D , Kutok JL , Schmidt-Supprian M , Rajewsky N and Yancopoulos G. Regulation of the germinal center response by microRNA-155. Science. 2007; 316: 604 -608. [PubMed] .
- 16. Lee RC , Feinbaum RL and Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75: 843 -854. [PubMed] .
- 17. Ambros V and Horvitz HR. Heterochronic mutants of the nematode Caenorhabditis elegans. Science. 1984; 226: 409 -416. [PubMed] .
- 18. Bonauer A , Carmona G , Iwasaki M , Mione M , Koyanagi M , Fischer A , Burchfield J , Fox H , Doebele C , Ohtani K , Chavakis E , Potente M and Tjwa M. MicroRNA-92a Controls Angiogenesis and Functional Recovery of Ischemic Tissues in Mice. Science. 2009; In press .
- 19. Ventura A , Young AG , Winslow MM , Lintault L , Meissner A , Erkeland SJ , Newman J , Bronson RT , Crowley D , Stone JR , Jaenisch R , Sharp PA and Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008; 132: 875 -886. [PubMed] .
- 20. Smibert P and Lai EC. Lessons from microRNA mutants in worms, flies and mice. Cell Cycle. 2008; 7: 2500 -2508. [PubMed] .
- 21. Alvarez-Garcia I and Miska EA. MicroRNA functions in animal development and human disease. Development. 2005; 132: 4653 -4662. [PubMed] .
- 22. Erson AE and Petty EM. MicroRNAs in development and disease. Clin Genet. 2008; 74: 296 -306. [PubMed] .
- 23. Bueno MJ , de Castro IP and Malumbres M. Control of cell proliferation pathways by microRNAs. Cell Cycle. 2008; 7: 3143 -3148. [PubMed] .
- 24. Chivukula RR and Mendell JT. Circular reasoning: microRNAs and cellcycle control. Trends Biochem Sci. 2008; 33: 474 -481. [PubMed] .
- 25. Jovanovic M and Hengartner MO. miRNAs and apoptosis: RNAs to die for. Oncogene. 2006; 25: 6176 -6187. [PubMed] .
- 26. Lu J , Getz G , Miska EA , Alvarez-Saavedra E , Lamb J , Peck D , Sweet- Cordero A , Ebert BL , Mak RH , Ferrando AA , Downing JR , Jacks T and Horvitz HR. MicroRNA expression profiles classify human cancers. Nature. 2005; 435: 834 -838. [PubMed] .
- 27. Chang TC , Yu D , Lee YS , Wentzel EA , Arking DE , West KM , Dang CV , Thomas-Tikhonenko A and Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008; 40: 43 -50. [PubMed] .
- 28. Bueno MJ , Perez de Castro I , Gomez de Cedron M , Santos J , Calin GA , Cigudosa JC , Croce CM , Fernandez-Piqueras J and Malumbres M. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008; 13: 496 -506. [PubMed] .
- 29. Calin GA , Dumitru CD , Shimizu M , Bichi R , Zupo S , Noch E , Aldler H , Rattan S , Keating M , Rai K , Rassenti L , Kipps T and Negrini M. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002; 99: 15524 -15529. [PubMed] .
- 30. Calin GA , Sevignani C , Dumitru CD , Hyslop T , Noch E , Yendamuri S , Shimizu M , Rattan S , Bullrich F , Negrini M and Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004; 101: 2999 -3004. [PubMed] .
- 31. Datta J , Kutay H , Nasser MW , Nuovo GJ , Wang B , Majumder S , Liu CG , Volinia S , Croce CM , Schmittgen TD , Ghoshal K and Jacob ST. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res. 2008; 68: 5049 -5058. [PubMed] .
- 32. Lujambio A , Calin GA , Villanueva A , Ropero S , Sanchez-Cespedes M , Blanco D , Montuenga LM , Rossi S , Nicoloso MS , Faller WJ , Gallagher WM , Eccles SA and Croce CM. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A. 2008; 105: 13556 -13561. [PubMed] .
- 33. Zhang L , Volinia S , Bonome T , Calin GA , Greshock J , Yang N , Liu CG , Giannakakis A , Alexiou P , Hasegawa K , Johnstone CN , Megraw MS and Adams S. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2008; 105: 7004 -7009. [PubMed] .
- 34. Migliazza A , Bosch F , Komatsu H , Cayanis E , Martinotti S , Toniato E , Guccione E , Qu X , Chien M , Murty VV , Gaidano G , Inghirami G and Zhang P. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001; 97: 2098 -2104. [PubMed] .
- 35. Migliazza A , Cayanis E , Bosch-Albareda F , Komatsu H , Martinotti S , Toniato E , Kalachikov S , Bonaldo MF , Jelene P , Ye X , Rzhetsky A , Qu X and Chien M. Molecular pathogenesis of B-cell chronic lymphocytic leukemia: analysis of 13q14 chromosomal deletions. Curr Top Microbiol Immunol. 2000; 252: 275 -284. [PubMed] .
- 36. Lee EJ , Baek M , Gusev Y , Brackett DJ , Nuovo GJ and Schmittgen TD. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. Rna. 2008; 14: 35 -42. [PubMed] .
- 37. Heo I , Joo C , Cho J , Ha M , Han J and Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell. 2008; 32: 276 -284. [PubMed] .
- 38. Newman MA , Thomson JM and Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. Rna. 2008; 14: 1539 -1549. [PubMed] .
- 39. Viswanathan SR , Daley GQ and Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008; 320: 97 -100. [PubMed] .
- 40. Viswanathan SR , Powers JT , Einhorn W , Hoshida Y , Ng TL , Toffanin S , O'Sullivan M , Lu J , Phillips LA , Lockhart VL , Shah SP , Tanwar PS and Mermel CH. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009; In press .
- 41. Suzuki HI , Yamagata K , Sugimoto K , Iwamoto T , Kato S and Miyazono K. Modulation of microRNA processing by p53. Nature. 2009; 460: 529 -533. [PubMed] .
- 42. Sandberg R , Neilson JR , Sarma A , Sharp PA and Burge CB. Proliferating cells express mRNAs with shortened 3' untranslated regions and fewer microRNA target sites. Science. 2008; 320: 1643 -1647. [PubMed] .
- 43. Kumar MS , Lu J , Mercer KL , Golub TR and Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007; 39: 673 -677. [PubMed] .
- 44. Mudhasani R , Zhu Z , Hutvagner G , Eischen CM , Lyle S , Hall LL , Lawrence JB , Imbalzano AN and Jones SN. Loss of miRNA biogenesis induces p19Arf-p53 signaling and senescence in primary cells. J Cell Biol. 2008; 181: 1055 -1063. [PubMed] .
- 45. Serrano M , Lin AW , McCurrach ME , Beach D and Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88: 593 -602. [PubMed] .
- 46. Christoffersen NR , Shalgi R , Frankel LB , Klausen M , Pilpel Y , Nielsen FC , Oren M and Lund AH. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2009; In press .
- 47. Johnson SM , Grosshans H , Shingara J , Byrom M , Jarvis R , Cheng A , Labourier E , Reinert KL , Brown D and Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005; 120: 635 -647. [PubMed] .
- 48. Sachdeva M , Zhu S , Wu F , Wu H , Walia V , Kumar S , Elble R , Watabe K and Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009; 106: 3207 -3212. [PubMed] .
- 49. Hanahan D and Weinberg RA. The hallmarks of cancer. Cell. 2000; 100: 57 -70. [PubMed] .
- 50. Olsen PH and Ambros V. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol. 1999; 216: 671 -680. [PubMed] .
- 51. Wightman B , Ha I and Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993; 75: 855 -862. [PubMed] .
- 52. Liu J , Carmell MA , Rivas FV , Marsden CG , Thomson JM , Song JJ , Hammond SM , Joshua-Tor L and Hannon GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004; 305: 1437 -1441. [PubMed] .
- 53. Meister G , Landthaler M , Patkaniowska A , Dorsett Y , Teng G and Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004; 15: 185 -197. [PubMed] .
- 54. Lim LP , Lau NC , Garrett-Engele P , Grimson A , Schelter JM , Castle J , Bartel DP , Linsley PS and Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005; 433: 769 -773. [PubMed] .
- 55. Baek D , Villen J , Shin C , Camargo FD , Gygi SP and Bartel DP. The impact of microRNAs on protein output. Nature. 2008; 455: 64 -71. [PubMed] .
- 56. Selbach M , Schwanhausser B , Thierfelder N , Fang Z , Khanin R and Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008; 455: 58 -63. [PubMed] .
- 57. Hornstein E and Shomron N. Canalization of development by microRNAs. Nat Genet. 2006; 38 Suppl: S20 -24. [PubMed] .
- 58. Brosh R , Shalgi R , Liran A , Landan G , Korotayev K , Nguyen GH , Enerly E , Johnsen H , Buganim Y , Solomon H , Goldstein I , Madar S and Goldfinger N. p53-Repressed miRNAs are involved with E2F in a feed-forward loop promoting proliferation. Mol Syst Biol. 2008; 4: 229 [PubMed] .
- 59. John B , Enright AJ , Aravin A , Tuschl T , Sander C and Marks DS. Human MicroRNA targets. PLoS Biol. 2004; 2: e363 [PubMed] .
- 60. Stark A , Brennecke J , Russell RB and Cohen SM. Identification of Drosophila MicroRNA targets. PLoS Biol. 2003; 1: E60 [PubMed] .
- 61. Shalgi R , Lieber D , Oren M and Pilpel Y. Global and Local Architecture of the Mammalian microRNA-Transcription Factor Regulatory Network. PLoS Comput Biol. 2007; 3: e131 [PubMed] .
- 62. Tsang J , Zhu J and van Oudenaarden A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell. 2007; 26: 753 -767. [PubMed] .
- 63. Milo R , Shen-Orr S , Itzkovitz S , Kashtan N , Chklovskii D and Alon U. Network motifs: simple building blocks of complex networks. Science. 2002; 298: 824 -827. [PubMed] .
- 64. Shen-Orr SS , Milo R , Mangan S and Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet. 2002; 31: 64 -68. [PubMed] .
- 65. Farh KK , Grimson A , Jan C , Lewis BP , Johnston WK , Lim LP , Burge CB and Bartel DP. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005; 310: 1817 -1821. [PubMed] .
- 66. Stark A , Brennecke J , Bushati N , Russell RB and Cohen SM. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3'UTR evolution. Cell. 2005; 123: 1133 -1146. [PubMed] .
- 67. Cohen EE , Zhu H , Lingen MW , Martin LE , Kuo WL , Choi EA , Kocherginsky M , Parker JS , Chung CH and Rosner MR. A feed-forward loop involving protein kinase Calpha and microRNAs regulates tumor cell cycle. Cancer Res. 2009; 69: 65 -74. [PubMed] .
- 68. Li X , Cassidy JJ , Reinke CA , Fischboeck S and Carthew RW. A microRNA imparts robustness against environmental fluctuation during development. Cell. 2009; 137: 273 -282. [PubMed] .
- 69. Marson A , Levine SS , Cole MF , Frampton GM , Brambrink T , Johnstone S , Guenther MG , Johnston WK , Wernig M , Newman J , Calabrese JM , Dennis LM and Volkert TL. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008; 134: 521 -533. [PubMed] .
- 70. O'Donnell KA , Wentzel EA , Zeller KI , Dang CV and Mendell JT. c-Myc regulated microRNAs modulate E2F1 expression. Nature. 2005; 435: 839 -843. [PubMed] .
- 71. Woods K , Thomson JM and Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. 2007; 282: 2130 -2134. [PubMed] .
- 72. Laurent LC , Chen J , Ulitsky I , Mueller FJ , Lu C , Shamir R , Fan JB and Loring JF. Comprehensive microRNA profiling reveals a unique human embryonic stem cell signature dominated by a single seed sequence. Stem Cells. 2008; 26: 1506 -1516. [PubMed] .
- 73. He L , Thomson JM , Hemann MT , Hernando-Monge E , Mu D , Goodson S , Powers S , Cordon-Cardo C , Lowe SW , Hannon GJ and Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005; 435: 828 -833. [PubMed] .
- 74. Li Y , Tan W , Neo TW , Aung MO , Wasser S , Lim SG and Tan TM. Role of the miR-106b-25 microRNA cluster in hepatocellular carcinoma. Cancer Sci. 2009; In press .
- 75. Mendell JT miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008; 133: 217 -222. [PubMed] .
- 76. Ivanovska I and Cleary MA. Combinatorial microRNAs: working together to make a difference. Cell Cycle. 2008; 7: 3137 -3142. [PubMed] .
- 77. Tanzer A and Stadler PF. Molecular evolution of a microRNA cluster. J Mol Biol. 2004; 339: 327 -335. [PubMed] .
- 78. He L , He X , Lim LP , de Stanchina E , Xuan Z , Liang Y , Xu W , Zender L , Magnus J , Ridzon D , Jackson AL , Linsley PS and Chen C. A microRNA component of the p53 tumour suppressor network. Nature. 2007; 447: 1130 -1134. [PubMed] .
- 79. Zhao Y , Samal E and Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005; 436: 214 -220. [PubMed] .
- 80. Chang TC , Wentzel EA , Kent OA , Ramachandran K , Mullendore M , Lee KH , Feldmann G , Yamakuchi M , Ferlito M , Lowenstein CJ , Arking DE , Beer MA and Maitra A. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007; 26: 745 -752. [PubMed] .
- 81. Raver-Shapira N , Marciano E , Meiri E , Spector Y , Rosenfeld N , Moskovits N , Bentwich Z and Oren M. Transcriptional activation of miR- 34a contributes to p53-mediated apoptosis. Mol Cell. 2007; 26: 731 -743. [PubMed] .
- 82. Tarasov V , Jung P , Verdoodt B , Lodygin D , Epanchintsev A , Menssen A , Meister G and Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007; 6: 1586 -1593. [PubMed] .
- 83. Kumamoto K , Spillare EA , Fujita K , Horikawa I , Yamashita T , Appella E , Nagashima M , Takenoshita S , Yokota J and Harris CC. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir- 34b, and mir-34c expression, and induce senescence. Cancer Res. 2008; 68: 3193 -3203. [PubMed] .
- 84. Kong YW , Cannell IG , de Moor CH , Hill K , Garside PG , Hamilton TL , Meijer HA , Dobbyn HC , Stoneley M , Spriggs KA , Willis AE and Bushell M. The mechanism of micro-RNA-mediated translation repression is determined by the promoter of the target gene. Proc Natl Acad Sci U S A. 2008; 105: 8866 -8871. [PubMed] .