


TAp63 maintains adult stem cells
The mysterious mechanisms that regulate aging are an area of active research. The induction of senescence or apoptosis in stem and progenitor cells is thought to trigger premature organismal aging [2]. Consistent with this idea, we found that the TAp63-/- mice had a significantly shortened life span compared to its wild-type littermates [1]. These mice exhibited phenotypes associated with premature aging including kyphosis, impaired wound healing, alopecia, epithelial and muscular atrophy, and chronic nephritis. These phenotypes suggest a critical role for TAp63 in the maintenance of adult stem cells in multiple epithelial and non-epithelial tissues. Indeed, we found that TAp63 maintains dermal stem cells by transcriptionally activating the cyclin dependent kinase inhibitor, p57, thereby preventing hyperproliferation of these cells (Figure 1A) [1,3]. Similar to the phenotype identified in dermal and epidermal progenitor and stem cells, other adult stem cells in the TAp63-/- mice may be hyperproliferative early in life and through similar senescence mechanisms that we delineated may result in a depletion of these stem cells and premature organismal aging (Figure 1B) [1].
The complex roles of the p53 family in aging
Increased p53 activity has been previously implicated in aging [4,5]. Although some mouse models with increased p53 activity exhibit signs of premature aging, others show conflicting results [6,7]. The important difference between these models is the alleles of p53 present in these mice. The mice exhibiting signs of premature aging contain truncated p53 mutants [4,5] while those that display a normal lifespan upregulate p53 by other mechanisms, such as the expression of a p53 transgene in addition to the endogenous p53 alleles or a hypomorphic allele of mdm2 [6,7]. One potential explanation of the discrepancy in the phenotypes of these mice is that TAp63 interacts with point mutant p53 rendering TAp63 functionally inactive. Consequently, mice expressing mutant p53 would exhibit phenotypes similar to those observed in the TAp63-/- mice. Previous studies have shown this to occur in the context of tumorigenesis and metastasis [8,9]. Mice engineered to express point mutants of p53 in Li-Fraumeni Syndrome inactivate p63 and p73 in tumors by binding to them and preventing the transactivation of their target genes [8,9,10]. These mouse models exhibit a metastatic phenotype similar to that observed in p53+/-;p63+/- and p53+/-;p73+/- mice illustrating an intricate relationship between the p53 family members [11,12].
Yet, another unexplored and possible explanation is that expression levels of the p53 family members change in mice that lack one or more of the family members, i.e. gene compensation. Such family member compensation has been observed in other families of genes including the Rb family [13,14,15]. In mouse models expressing abnormally high levels of p53, TAp63 levels may be dampened commensurate with an increase in p53 protein expression. p53 protein levels are known to be high in mice expressing mutated versions of p53 [8,9,10]. Thus, loss of TAp63 in these mouse models may again result in an acceleration of organismal aging. Furthermore, other isoforms of p63 and p73 have been implicated in premature aging [16,17]. Therefore, careful characterization of the expression of the other p53 family members, including the individual isoforms of p63 and p73, is necessary in mouse models expressing altered levels of p53 in order to understand the complex interplay and potential compensation between the p53 family members in processes that regulate longevity (Figure 1).
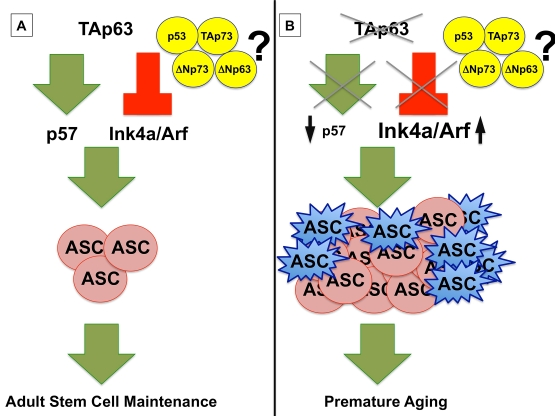
Figure 1. (A) TAp63
maintains adult stem cells (ASC) by transcriptionally activating p57
and repressing Ink4a/Arf, preventing premature aging. (B)
In the absence of TAp63, p57 mRNA levels are low, leading to
hyperproliferation of ASCs (shown in pink), and Ink4a/Arf levels are
high, resulting in a concomitant senescence of ASCs (shown in blue) and a
premature aging phenotype in TAp63 deficient mice. The interplay of
the p53 family, including TAp73, ΔNp73, and ΔNp63, remains to be
elucidated.
Loss of TAp63 triggers senescence and cannot be reversed by concomitant loss of p53
Interestingly and surprisingly, senescence triggered in TAp63-/- epidermal precursors is p53-independent. In fact, we found a higher proportion of senescent cells in TAp63-/-;p53-/- epidermal cells than in those lacking TAp63 only, indicating that loss of p53 does not bypass senescence in this tissue [1]. This further indicates that TAp63 directly regulates senescence in epidermal precursor cells by transcriptionally repressing Ink4a and Arf as has been observed in the epidermis of mice deficient for p63 [1,18]. The mechanisms employed by TAp63 to induce senescence have important implications for deciphering its role as a tumor suppressor gene.
TAp63 is induced in response to stress
p63 evolved to have several isoforms that can be divided into two categories: the TA (transactivation competent isoforms) and the ΔN (those that lack the transactivation domain). The most highly expressed isoforms of p63 in the skin are the ΔNp63 isoforms, thus the prevailing view is that ΔNp63, and more specifically ΔNp63α, are the isoforms that play critical roles in maintaining the epidermis [19,20]. However, it is important to note that the TAp63 isoforms structurally resemble p53 and have been shown in other systems to be induced in response to DNA damage and stress [21,22]. Importantly, although TAp63 protein expression is undetectable in the normal epidermis, we found that TAp63 expression increased dramatically in response to stress induced by wounding, indicating that much like p53, TAp63 serves to protect cells from damage [1]. This is a novel and unrecognized role for TAp63 in maintaining the dermis and the integrity of the epidermis.
TAp63: The key to longevity?
Mice lacking TAp63 also develop severe skin erosions that do not heal [1]. These erosions result from trauma or ruptured blisters that form in the majority of TAp63-/- mice. The failure of these mice to appropriately heal their wounds results from a depletion of SKP cells known to be required for wound healing [1]. Additionally, the TAp63-/- mice exhibited patches where there was a diminution in the number of hair follicles resulting in alopecia in these mice. Some of these defects are similar to those seen in patients with Hay-Wells syndrome or ankyloblepharon-ectodermal dysplasia-clefting (AEC) syndrome [23]. These pa-tients develop alopecia and skin erosions with impaired wound healing indicating that the TAp63-/- mouse may be useful as a preclinical model to test therapies for these disfiguring and painful diseases.
In addition, given the critical function of TAp63 in wound healing and hair growth, reactivation of TAp63 in tissues of patients with degenerative diseases has important therapeutic implications not only in patients with AEC syndrome but also in those with impaired wound healing, like diabetes. Important areas for future investigation include developing models and therapies whereby TAp63 can be reactivated in adult dermal stem cells to determine whether senescence and premature aging can be reversed in these cells to aid in the wound healing process and hair regeneration.
The impact of the TAp63-/- aging phenotype on cancer
p63 is an important suppressor of tumorigenesis and metastasis; however, at first glance, the role of p63 in senescence and aging may seem at odds with its role as a tumor suppressor. It is important to note that adult dermal stem cells are initially hyperproliferative prior to acquiring a senescent phenotype (Figure 1B). By extension, in tumor formation, cancer stem or precursor cells that lose TAp63 may likewise be hyper-proliferative. With the high levels of DNA damage and genomic instability that are detected in dermal and epidermal stem cells lacking TAp63 [1], these cancer stem cells will likely acquire new mutations that allow escape from senescence, an ideal formula for tumor formation. In addition to further investigation on how TAp63 affects cancer stem cells, the milieu in which cancer cells reside must also be closely examined in the TAp63-/- mouse model. Cancer incidence increases with age, and it is possible that the prematurely aged environment of the TAp63-/- mouse provides an ideal environment for tumor formation and metastasis. Further investigation on the effects of premature aging in the TAp63 deficient mouse model on tumor formation is critical to obtain an understanding of the roles of TAp63 as a tumor suppressor gene.
In summary, we have revealed a critical role for TAp63in preventing premature aging and further complexity of the p53 family, underscoring a need to understand the family as a whole and its roles in human diseases. A clear understanding of the intimate and complex relationship between the p53 family of genes is essential to target this pathway in degenerative diseases and tumorigenesis.
Acknowledgments
E.R.F. is a scholar of the American Cancer Society (RSG-07-082-01-MGO), Rita Allen Foundation, V Foundation for Cancer Research, and March of Dimes (Basil O'Connor Scholar). We thank Kenneth Y. Tsai for critical reading of the manuscript.
Conflicts of Interest
The authors have no conflict of interests to declare.
References
- 1. Su X , Paris M , Gi YJ , Tsai KY , Cho MS , Lin YL , Biernaskie JA , Sinha S , Prives C , Pevny LH , Miller FD and Flores ER. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009; 5: 64 -75. [PubMed] .
- 2. Finkel T , Serrano M and Blasco MA. The common biology of cancer and ageing. Nature. 2007; 448: 767 -774. [PubMed] .
- 3. Beaudry VG and Attardi LD. SKP-ing TAp63: stem cell depletion, senescence, and premature aging. Cell Stem Cell. 2009; 5: 1 -2. [PubMed] .
- 4. Maier B , Gluba W , Bernier B , Turner T , Mohammad K , Guise T , Sutherland A , Thorner M and Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004; 18: 306 -319. [PubMed] .
- 5. Tyner SD , Venkatachalam S , Choi J , Jones S , Ghebranious N , Igelmann H , Lu X , Soron G , Cooper B , Brayton C , Hee Park S , Thompson T and Karsently G. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002; 415: 45 -53. [PubMed] .
- 6. Garcia-Cao I , Garcia-Cao M , Martin-Caballero J , Criado LM , Klatt P , Flores JM , Wells JC , Blasco MA and Serrano M. "Super p53" mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002; 21: 6225 -6235. [PubMed] .
- 7. Mendrysa SM , O'Leary KA , McElwee MK , Michalowski J , Eisenman RN , Powell DA and Perry ME. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006; 20: 16 -21. [PubMed] .
- 8. Lang GA , Iwakuma T , Suh YA , Liu G , Rao VA , Parant JM , Valentin-Vega YA , Terzian T , Caldwell LC , Strong LC , El-Naggar AK and Lozano G. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004; 119: 861 -872. [PubMed] .
- 9. Olive KP , Tuveson DA , Ruhe ZC , Yin B , Willis NA , Bronson RT , Crowley D and Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004; 119: 847 -860. [PubMed] .
- 10. Iwakuma T , Lozano G and Flores ER. Li-Fraumeni syndrome: a p53 family affair. Cell Cycle. 2005; 4: 865 -867. [PubMed] .
- 11. Flores ER The roles of p63 in cancer. Cell Cycle. 2007; 6: 300 -304. [PubMed] .
- 12. Flores ER , Sengupta S , Miller JB , Newman JJ , Bronson R , Crowley D , Yang A , McKeon F and Jacks T. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005; 7: 363 -373. [PubMed] .
- 13. Mulligan G and Jacks T. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet. 1998; 14: 223 -229. [PubMed] .
- 14. Mulligan GJ , Wong J and Jacks T. p130 is dispensable in peripheral T lymphocytes: evidence for functional compensation by p107 and pRB. Mol Cell Biol. 1998; 18: 206 -220. [PubMed] .
- 15. Sage J , Miller AL , Perez-Mancera PA , Wysocki JM and Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003; 424: 223 -228. [PubMed] .
- 16. Keyes WM , Wu Y , Vogel H , Guo X , Lowe SW and Mills AA. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005; 19: 1986 -1999. [PubMed] .
- 17. Wetzel MK , Naska S , Laliberte CL , Rymar VV , Fujitani M , Biernaski JA , Cole CJ , Lerch JP , Spring S , Wang SH , Frankland PW , Henkelman RM and Josselyn SA. p73 regulates neuro-degeneration and phospho-tau accumulation during aging and Alzheimer's disease. Neuron. 2008; 59: 708 -721. [PubMed] .
- 18. Su X , Cho MS , Gi YJ , Ayanga BA , Sherr CJ and Flores ER. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009; 28: 1904 -1915. [PubMed] .
- 19. Candi E , Rufini A , Terrinoni A , Dinsdale D , Ranalli M , Paradisi A , De Laurenzi V , Spagnoli LG , Catani MV , Ramadan S , Knight RA and Melino G. Differential roles of p63 isoforms in epidermal development: selective genetic complementation in p63 null mice. Cell Death Differ. 2006; 13: 1037 -1047. [PubMed] .
- 20. Senoo M , Pinto F , Crum CP and McKeon F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007; 129: 523 -536. [PubMed] .
- 21. Flores ER , Tsai KY , Crowley D , Sengupta S , Yang A , McKeon F and Jacks T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002; 416: 560 -564. [PubMed] .
- 22. Jacobs WB , Govoni G , Ho D , Atwal JK , Barnabe-Heider F , Keyes WM , Mills AA , Miller FD and Kaplan DR. p63 is an essential proapoptotic protein during neural development. Neuron. 2005; 48: 743 -756. [PubMed] .
- 23. McGrath JA , Duijf PH , Doetsch V , Irvine AD , de Waal R , Vanmolkot KR , Wessagowit V , Kelly A , Atherton DJ , Griffiths WA , Orlow SJ , van Haeringen A and Ausems MG. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001; 10: 221 -229. [PubMed] .