



Aging
In human biology aging is accompanied by a diminished capacity to adequately maintain tissue homeostasis or to repair tissues after injury. When homeostatic control diminishes to a point at which tissue/organ integrity and function are no longer sufficiently maintained, physiologic decline ensues, and aging is manifested. Consistent with this, many of the pathophysiological conditions afflicting the elderly, such as anemia, sarcopenia and osteoporosis, suggest an imbalance between cell loss and renewal.
Numerous theories have been put forth to explain the decline of stem cell function with advancing age. The free-radical theory of aging proposes that reactive oxygen species (ROS), which are by-products of normal metabolism, are responsible for damage to many cellular components, including DNA [1]. It is clear, however, that in addition to ROS, a much broader range of extrinsic and intrinsic sources, such as UV irradiation, alkylating agents, telomere attrition, and DNA replication errors, can also infringe upon genomic integrity [2]. In response to this damage, tumor suppressor pathways are activated, including those mediated by the tumor suppressor proteins p16 and p53, to ensure that potentially dangerous lesions do not lead to malignancy [2,3]. So, when aging advances and damage accumulates the activity of these tumor suppressors increases and consequently has the potential to negatively modulate stem cell function through the induction of apoptosis or senescence.
p16INK4a and aging
The cyclin-dependent kinase inhibitor p16INK4a has emerged as an important player in aging and age-related disease. p16INK4a has the ability to bind and inhibit the cyclin-D-dependent kinases CDK4 and CDK6. These kinases are known to have oncogenic potential and phosphorylate the retinoblastoma (Rb) family of tumor suppressors. Hence, expression of p16INK4a maintains Rb-family proteins in hypophosphorylated state, which promotes binding of E2F to effect a G1 cell cycle arrest [3,4]. Several studies have shown that p16INK4a expression increases markedly with advancing age in a variety of tissues [5-8]. Consequently, the increased expression of p16INK4a induces an age-dependent decrease in the proliferative capacity of certain tissue-specific stem- and progenitor-cells [5-8]. Senescence characterized by p16INK4a upregulation as well as telomere shortening has been observed in human and mouse cardiomyocytes and may contribute to myocardial aging [9,10]. Because p16INK4a expression can be upregulated by a wide variety of stresses [4,11] it may be involved in many, maybe all, forms of senescence, and has thus recently received attention as a promising biomarker [12,13].
Also in the hematopoietic system it has been shown that the induction of p16INK4a correlated with the in vivo senescence of hematopoietic stem cells [14-16]. Indeed, mice lacking BMI1, a repressor of p16INK4a, displayed a striking loss of hematopoietic cells which correlated with increased expression of p16INK4a expression and replicative failure of stem cells [16-18]. Conversely, an increase in regenerative capacity was found in the bone marrow of p16INK4a deficient mice [19]. These findings reinforce the notion that the age-associated upregulation of p16INK4a restricts self-renewal and unbalances tissue homeostasis. We have confirmed increased p16INK4a expression in healthy human CD34+ hematopoietic cells upon aging [20].
Aging and Acute Myeloid Leukemia
Aging not only affects normal hematopoietic development (e.g. increased incidence of anemia), but also impacts on the clinical biology of AML. In particular, the incidence of AML increases with increasing age [21,22]. Moreover, older AML patients have a markedly reduced long-term survival due to the combination of poor chemotherapeutic tolerance and inherent chemotherapy resistance compared to younger AML patients [21-25]. AML in older patients shows also a lower frequency of favorable core-binding chromosomal abnormalities and a higher incidence of complex aberrant karyotypes [26,27]. We wondered whether these differences in clinical and cellular behavior of AML in older patients were reflected by differences in gene expression profiles. Therefore, a cohort of 525 adult AML patients was studied to compare gene expression profiles of the one-third of youngest cases (median age 31 years) with the one third of oldest cases (median age 59 years) [20]. Biological processes (represented by GO-ontologies) associated with aging in AML were compared with a published list of significantly differentially expressed GO-ontologies between young and old purified murine long-term hematopoietic stem cells. This analysis revealed that in both sets the NF-κB cascade was up-regulated and that maintenance of chromatin architecture, chromatin modification and organelle organization were down-regulated [20, 28]. Subsequently, comparison of gene expression profiles of AML samples of the 175 youngest with the 175 oldest AML patients revealed that 477 probe sets were up-regulated and 492 probe sets were down-regulated with increasing age at the significance level of P <1.0x10-5. Additionally, two in- dependent AML gene array cohorts were used for validation. The final list of validated genes which were differently expressed depending on age in three independent AML cohorts yielded a number of interesting genes, including p16INK4a. The level of expression of p16INK4a in AML samples during aging was reciprocal to the usual trend of an increased expression at higher age; i.e. the expression of p16INK4a declined significantly with increasing age. Of note, this was only noticed in the intermediate- and unfavorable-risk group and not in the favorable-risk group and the molecularly defined subset ‘NPM1 mutant without FLT3-ITD'. Multivariate analysis revealed p16INK4a, besides cytogenetic risk-groups, as an independent prognostic parameter for overall survival (OS) in older (and not in younger) AML patients. Lower p16INK4a expression in AML samples of patients of older age predicts for reduced OS. Further studies unraveling the regulation and molecular mechanisms responsible for the down-regulation of p16INK4a during aging in AML are awaited. Striking in this perspective is our observation that BMI1, a potent repressor of p16INK4a, was significantly inversely correlated with p16INK4a in older (P = <.001, rho = -.274, n = 175) and not in younger (P = .322, rho = -.075, n = 175) AML patients.
Is the downregulation of p16INK4a a broader phenomenon?
We wondered whether the paradoxical down-regulation of p16INK4a with advancing age in AML might be a more general phenomenon, i.e. is p16INK4a expression also down-regulated with advancing age in other malignancies? An extensive search in the Gene Expression Omnibus (GEO) repository revealed only three publically available Affymetrix gene array datasets of malignancies (i.e. lymphoma, glioblastoma and breast cancer) which also presented the patient characteristic age (GSE4475, GSE7696 and GSE3494) [29-31]. The median age at diagnosis is given in Table 1 for all three cohorts. Interestingly, the expression of p16INK4a declined significantly with advancing age in lymphoma patient samples (P = <.001, rho = -.252 n = 219, Table 1) and in glioblastoma patient samples (P = .005, rho = -.310, n= 80, Table 1). In breast cancer patient samples the continuous variables age and p16INK4a did not correlate (P = .407, rho = -.053, n = 251, Table 1).
Table 1. Correlation between the expression of p16 INK4a with age with age at diagnosis in three malignancies.
The relation between the expression of p16INK4a mRNA level and age at diagnosis in three malignancies (publicly available micro arrays, i.e. GSE4475, GSE7696 and GSE3494)[29-31]. Spearman rank correlation coefficients between the continuous variables age and the averaged p16INK4a probe sets (n=3) were calculated. The characteristic age is given as median (range).
| Cancer type | age | n | P | Rho |
| lymphoma | 61 (2-61) | 219 | <.001 | -.252 |
| glioblastoma | 52 (26-70) | 80 | .005 | -.310 |
| breast cancer | 64 (28-93) | 251 | .407 | -.053 |
Conclusive Remarks
The cyclin-dependent kinase inhibitor p16INK4a, which has emerged as an important effector of aging and a potent tumor suppressor, was shown to be down-regulated in older AML samples [20]. However, this is not unique for AML since the paradoxical down-regulation with advancing age is also observed in samples from lymphoma and glioblastoma patients. These observations provide an interesting new link between aging and cancer. We hypothesize that suppression of defense mechanisms which protect older cells against cellular and DNA damage might facilitate oncogenesis in cancer cells from older individuals (Figure 1). Accordingly, the interaction between aging and cancer warrants further studies, to determine whether age-associated protection mechanisms play a specific role in cancer of older patients and to determine whether modulation of these age-associated protection mechanisms can be exploited in the treatment of older cancer patients.
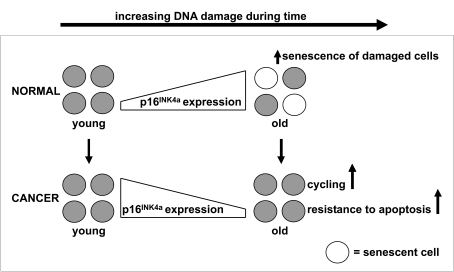
Figure 1. p16 INK4a expression during aging of healthy and malignant cells.
The expression of p16INK4a mRNA increases with advancing age to
ensure that potentially dangerous lesions, due to accumulated DNA damage,
do not lead to malignancy. The increased expression of p16INK4a
mRNA has the potential to negatively modulate stem cell function through
the induction of apoptosis or senescence. Our data illustrate the
importance of this p16INK4a dependent mechanism, since samples
of older cancer patients have a lower instead of higher expression of p16INK4a
mRNA compared to samples of younger cancer patients. So, we hypothesize
that suppression of defense mechanisms which protect older stem cells
against accumulated cellular and DNA damage facilitates the development of
cancer in older individuals.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Finkel T and Holbrook NJ. Oxidants, oxidative stress and the biology of aging. Nature. 2000; 408: 239 -246. [PubMed] .
- 2. Rossi DJ , Jamieson CHM and Weissman IL. Stem cells and the pathways to aging and cancer. Cell. 2008; 132: 681 -696. [PubMed] .
- 3. Collado M , Blasco MA and Serrano M. Cellular senescence in cancer and aging. Cell. 2007; 130: 223 -233. [PubMed] .
- 4. Kim WY and Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006; 127: 265 -275. [PubMed] .
- 5. Krishnamurthy J , Torrice C , Ramsey MR , Kovalev GI , Al Regaiey K , Su L and Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004; 114: 1299 -1307. [PubMed] .
- 6. Melk A , Schmidt BM , Takeuchi O , Sawitzki B , Rayner DC and Halloran PF. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004; 65: 510 -520. [PubMed] .
- 7. Nielsen GP , Stemmer-Rachamimov AO , Shaw J , Roy JE , Koh J and Louis DN. Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab Invest. 1999; 79: 1137 -1143. [PubMed] .
- 8. Zindy F , Quelle DE , Roussel MF and Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997; 15: 203 -211. [PubMed] .
- 9. Chimenti C , Kajstura J , Torella D , Urbanek K , Heleniak H , Colussi C , Di Meglio F , Nadal-Ginard B , Frustaci A , Leri A , Maseri A and Anversa P. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res. 2003; 93: 604 -13. [PubMed] .
- 10. Torella D Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004; 94: 514 -24. [PubMed] .
- 11. Gil J and Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006; 7: 667 -677. [PubMed] .
- 12. Michaloglou C , Vredeveld LC , Soengas MS , Denoyelle C , Kuilman T , van der Horst CM , Majoor DM , Shay JW , Mooi WJ and Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005; 436: 720 -724. [PubMed] .
- 13. Ressler S , Bartkova J , Niederegger H , Bartek J , Scharffetter-Kochanek K , Jansen-Dürr P and Wlaschek M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006; 5: 379 -389. [PubMed] .
- 14. Lewis JL , Chinswangwatanakul W , Zheng B , Marley SB , Nguyen DX , Cross NC , Banerji L , Glassford J , Thomas NS , Goldman JM , Lam EW and Gordon MY. The influence of INK4 proteins on growth and self-renewal kinetics of hematopoietic progenitor cells. Blood. 2001; 97: 2604 -2610. [PubMed] .
- 15. Meng A , Wang Y , Van Zant G and Zhou D. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res. 2003; 63: 5414 -5419. [PubMed] .
- 16. Park I , Qian D and Kiel M. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003; 423: 302 -305. [PubMed] .
- 17. Lessard J and Sauvageau G. Bmi-1 determines the proliferative capacity in normal and leukaemic stem cells. Nature. 2003; 423: 255 -260. [PubMed] .
- 18. Molofsky AV , Pardal R , Iwashita T , Park IK , Clarke MF and Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003; 425: 962 -967. [PubMed] .
- 19. Janzen V , Forkert R and Fleming HE. Stem-cell aging modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006; 443: 421 -426. [PubMed] .
- 20. de Jonge HJ , de Bont ES , Valk PJ , Schuringa JJ , Kies M , Woolthuis CM , Delwel R , Veeger NJGM , Vellenga E , Löwenberg B and Huls G. AML at older age: age related gene expression profiles reveal a paradoxical down-regulation of p16INK4A mRNA with prognostic significance. Blood. 2009; 114: 2869 -287721. [PubMed] .
- 21. Appelbaum FR , Gundacker H and Head DR. Age and acute myeloid leukemia. Blood. 2006; 107: 3481 -3485. [PubMed] .
- 22. Frohling S , Schlenk RF and Kayser S. Cytogenetics and age are major determinants of outcome in intensively treated acute myeloid leukemia patients older than 60 years: results from AMLSG trial AML HD98-B. Blood. 2006; 108: 3280 -3288. [PubMed] .
- 23. Estey E Acute myeloid leukemia and myelodysplastic syndromes in older patients. J Clin Oncol. 2007; 25: 1908 -1915. [PubMed] .
- 24. Grimwade D , Walker H and Harrison G. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001; 98: 1312 -1320. [PubMed] .
- 25. Hiddemann W , Kern W and Schoch C. Management of acute myeloid leukemia in elderly patients. J Clin Oncol. 1999; 17: 3569 -3576. [PubMed] .
- 26. Rowley JD , Alimena G , Garson OM , Hagemeijer A , Mitelman F and Prigogina EL. A collaborative study of the relationship of the morphological type of acute nonlymphocytic leukemia with patient age and karyotype. Blood. 1982; 59: 1013 -1022. [PubMed] .
- 27. Schoch C , Kern W and Krawitz P. Dependence of age-specific incidence of acute myeloid leukemia on karyotype. Blood. 2001; 98: 3500 [PubMed] .
- 28. Chambers SM , Shaw CA , Gatza C , Fisk CJ , Donehower LA and Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007; 5: e201 [PubMed] .
- 29. Hummel M A biologic definition of Burkitt's lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006; 354: 2419 -2430. [PubMed] .
- 30. Miller LD , Smeds J , George J , Vega VB , Vergara L , Ploner A , Pawitan Y , Hall P , Klaar S , Liu ET and Bergh J. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci. 2005; 102: 13550 -13555. [PubMed] .
- 31. Murat A Stem cell-related "self-renewal" signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblasto-ma. J Clin Oncol. 2008; 26: 3015 -3024. [PubMed] .