



Culture shock causes rapid replicative senescence in primary MEFs with functional p53, but p53 deficient cells are resistant
Cellular senescence is an important defence mechanism against tumour metastasis, growth and progression [1,2]. Furthermore, in both humans and in mouse models, cancers can respond to chemotherapy by a massive senescence response followed by tumour cell clearance [3-5]. MEFs are a classic model system for studying cell senescence and immortalisation, with clear parallels to key genetic alterations during human tumourigenesis whilst offering expedient advantages over human cell cultures when exploring basic molecular mechanisms of senescence control and senescence bypass [6,7]. When explanted in vitro MEFs initially continue to replicate, but then rapidly undergo stress-associated senescence due to in vitro conditions, especially oxidative stress elicited by standard culturing conditions, which supply supraphysiological levels of oxygen [8]. DNA damage from reactive oxygen species is clearly a key factor in this senescence response [8,9]. As is well known from research in several laboratories, cells that sustain spontaneous damage to the p53/p19ARF pathway (p14 in humans) can overcome this replication block, leading to gradual outgrowth of an immortalised cell population with unlimited growth potential [10-14].
Our MEF cell line library comprises immortalised cell lines from cultures of fibroblasts derived from (a) embryos of a standard laboratory wild-type mouse strain (129/Sv), and (b) embryos from human p53 knock-in (Hupki) mice, which we constructed for our research on p53 biology [15-17]. The original Hupki mouse strain [15] harbours normal human p53 gene sequences encoding the DNA binding domain and the polyproline domain embedded in the endogenous murine p53 locus. This strain is phenotypically normal, not tumour-prone, and displays classical p53 wild-type responses, including DNA damage induced apoptosis, transcriptional transactivation of p53 target genes and stress-induced cellular senescence. Curiously, a mouse strain in which the entire p53 sequence was replaced by the human counterpart lost wild-type p53 function due to abnormal interactions with the p53 negative regulator Mdm2 [18]. The Hupki strain is not p53-deficient, and can be used as a source of primary MEFs, thus allowing the extensive literature on MEF senescence bypass, and the database of human tumour suppressor mutations to be linked to specific mutations that support senescence bypass. Using this approach, we have shown that the basic features of stress-induced senescence and immortalisation are comparable in MEFs from standard strain wild-type (WT) mice and Hupki mice. Studies from our laboratory with several hundred immortalised Hupki MEF cell lines have shown that the genetic alterations in p53 that lead to senescence bypass of MEFs are typical of human tumours. Missense point mutations in p53 and p19/ARF silencing by biallelic deletion, also common in human tumours, are the two most common routes to spontaneous p53/p19ARF pathway inactivation in immortalised MEFs identified thus far ([16,19,20] and unpublished observations).
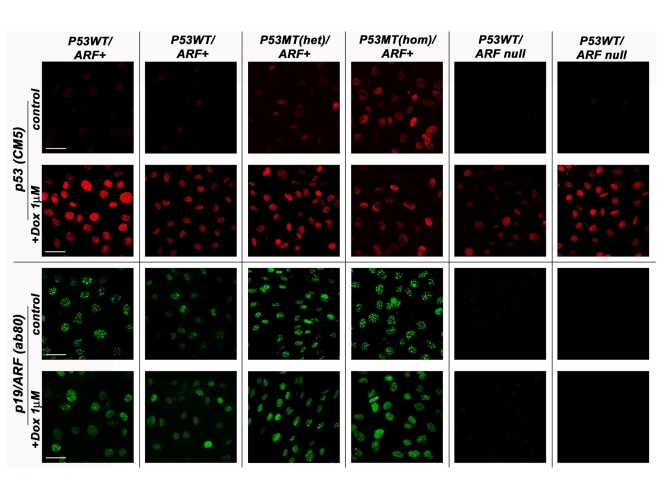
Figure 1. The p53/p19ARF status of various MEF lines derived from normal strain 129 mice. MEF cell lines
genotyped as either wild-type (WT) p53 or mutant (MT) p53 (both
heterozygous and homozygous) were compared against those carrying the
p19/ARF deletion in their response to doxorubicin (8h, 1μM) treatment.
Cells were methanol fixed and processed by indirect immunofluorescence
confocal microscopy with either the anti-p53 CM5 (Novacastra) or
anti-p19/ARF ab80 (AbCam) antibody. Scale bar represents 50μm. All samples
were processed at the same intensity and magnification.
Whilst the prevalence of cell lines immortalised by p19/ARF biallelic deletion or p53 mutation was not unexpected (up to 50 % of cell lines, depending on immortalisation protocols), we were surprised to find that a significant fraction of cell lines (derived both from WT and Hupki primary cells) appeared to have retained WT p53 and p19/ARF expression, as examined by DNA sequencing or PCR amplification and promoter methylation analyses, respectively ([20] and unpub-lished). Probing by immunofluorochemistry for p53 nuclear accumulation and induction of p21/WAF1 following exposure of these cell lines to the DNA damaging agent doxorubicin (Figure 1), as well as detection of p19/ARF protein by immunoblotting [20] support the p53, p19/ARF wild-type status of these cell lines. What might then be the genetic alterations that allowed these cells to bypass senescence? One possibility concerns the phenomenon of tumour suppressor haploinsufficiency in promoting cell growth (see insightful review by Quon and Berns, 2001) [21,22]. Contrary to the original 2-hit paradigm for tumour suppressor genes, where both alleles must be inactivated to elicit a growth promoting phenotype, ample evidence from in vivo and in vitro studies and human tumour analyses demonstrates that not only the absence, but also the moderate reduction of tumour suppressor gene products can be sufficient to alter growth phenotype. In the case of MEFs, as expected, cells from p53 null mice fail to senesce when explanted in vitro under standard culture conditions, but cells from heterozygous Hupki mice (progeny of Hupki and p53 null mice) also continue to grow when explanted, with only a brief slowing of doubling time after the first several passages (unpublished observations). Some of these immortal cultures eventually do reveal loss of the WT allele with continued passaging, but others retain the original unmutated p53 allele. This suggests that the presence of only one WT allele in primary explanted MEFs may be sufficient to bypass senescence initially. Conceivably, as passage number increases, so will the chances that the WT allele is eventually discarded, possibly provoking a jump in growth rate, as we have noted for some slow-growing MEF cell lines.
Some of the MEF cell lines from (homozygous) WT or Hupki mice that we have examined and tentatively classified as p53 and p19/ARF wild-type thus may in fact have only one normal allele of these suppressor genes, having suffered loss of one copy in vitro allowing unlimited growth in culture.
Beyond p53/p19ARF
Given that at least half of the spontaneously arising >100 MEF cell lines we examined for p53 and p19/ARF aberrations appeared to have overcome senescence block upon explanting in vitro by mechanisms other than the 2 canonical genetic events (p53 mutation; p19 biallelic deletion), there could well be various alternative pathways to immortalisation not directly involving damage to the immediate p53/p19ARF axis.
Considerable effort is underway to identify the key regulators of senescence and immortalisation. Since the process of senescence bypass provides an intrinsic phenotypic readout of functionality and automatically generates cell lines amenable to subsequent analysis, reverse genetics is a powerful approach to deciphering the important molecular events involved in senescence control [23]. Several large-scale screens for genes involved in senescence/senescence bypass have been performed, including both gain-of-function screens involving the ectopic expression of cDNA libraries as well as loss-of-function screens involving the expression of antisense cDNA libraries and shRNA libraries. These screens have been performed in human and mouse cell models of both replicative and oncogene-induced senescence and each approach has identified different genes involved in senescence, providing novel and sometimes unexpected insights into the process (Table 1). Reassuringly, well established players in the master regulatory pathways of senescence (for example p53) have also been identified in these screens [24,25]. Indeed, inactivation of p53 is often used as a positive control in such experiments [24,26]. While p53 knock-down in senescent MEFs has been shown to reverse senescence [27], this may or may not be true of other genes involved in senescence which may require inactivation or expression prior to the acquisition of senescence.
The role of p53 mutation/p19ARF deletion in senescence bypass in MEFs reflects the importance of the p53/p19ARF axis as a master regulatory pathway of senescence in these cells. What is apparent from genetic screening, as well as from other complementary work, is that many of the novel senescence-associated genes identified can also impact on this key pathway, both upstream and downstream of p53 [24,26,28-31]. The p16/pRb pathway, another senescence master regulator, is also commonly affected by novel senescence-associated molecules [28,32]. A number of additional interacting signaling pathways have been implicated in the induction or bypass of senescence including the RAS/MAPK pathway [25,33,34], the AKT pathway [35-37] and the JNK pathway [38,39], although the relative contribution of these to the senescent phenotype appears to be dependent on species, cell type and the pro-senescence stimulus.
Concluding remarks: Senescence - good or bad?
An entirely new aspect to the importance of cellular senescence has recently surfaced from experiments to produce iPS (induced pluripotent stem) cells from embryonic fibroblasts. It has been shown that senescence provides a progressive barrier to conversion of primary MEFs (and indeed other types of differentiated cells) to pluripotency. Crucially, disruption of the p53/p19ARF signalling axis greatly increased efficiency of their conversion [40,41]. Furthermore, genetic ablation of p53 in cells normally considered refractory to reprogramming into pluripotent stem cells can overcome this block [40]. Clearly, senescence is a key process to target in optimising strategies to enhance somatic cell reprogramming. Identification of factors influencing senescence will reveal novel genes/pathways to modulate that could enhance conversion to pluripotency without compromising genetic integrity, expediting potential applications for iPS cells in regenerative medicine.
Table 1. Novel genes identified in reverse-genetics senescence bypass screens.
The table shows the diversity of genes which either promote senescence or its bypass as identified in cellular screens for senescence bypass. Genes well known to be important in cellular senescence such as p53, p21 and PAI-1 are not included here. aOther genes identified in this screen: BNIP3L, BIN1, HSPA9, IL1R1, PEA15, RAP1GAP, DMTF1, FOXA1, IRF1, MEN1, HIRA, SMARCB1, FBXO31, NF2 [25]. bAdditional genes identified in this screen: RPS6KA6, HTATIP, HDAC4, SAH3, CCT2 [24].
| Gene | Promotion or inhibition of senescence | Potential senescence-associated pathway/mechanism of action | Biological Function | Refe-rence | Cell type |
| BCL6 | inhibition | Induces cyclin D1 expression and renders cells unresponsive to antiproliferative signals from the p19(ARF)-p53 pathway | Transcription factor | [29] | MEFs, human B cells |
| Bub1 | inhibition | Bub1 RNAi induces senescence. Bub1 expression does not extend lifespan | Mitotic checkpoint Ser/Thr kinase | [25a, 28, 42] | Primary MEFs |
| Csn2 | promotion | Inactivation inhibits p53 transcriptional activity and confers resistance to both p53- and p16INK4a-induced proliferation arrest | Component of the Cop9 signalosome | [28] | Primary MEFs |
| Brf1 | promotion | Inhibition of p53 transcription and reduction p16ink4a-induced arrest | Subunit of the RNA polymerase II complex | [28] | Primary MEFs |
| Aldose Reductase | promotion | Inhibition of p53 transcription and reduction p16ink4a-induced arrest | Metabolic enzyme - glucose metabolism | [28] | Primary MEFs |
| Tid1 | induction | Tid1 is a repressor of NF-κB signaling | DNA-J like protein which functions as a co-chaperone | [43] | Rat embryo fibroblasts |
| hDRIL1 | inhibition | Renders primary MEFs unresponsive to RAS(V12)-induced anti-proliferative signaling by p19(ARF)/p53/p21(CIP1), as well as by p16(INK4a) Binds E2F1 and induces Cyclin E1 | Transcription factor | [32] | MEFs RASV12 induced senescence |
| CBX7 | inhibition | Controls cellular lifespan through regulation of both the p16(Ink4a)/Rb and the Arf/p53 pathways Represses INK4a-ARF locus | Transcription factor | [44] | Normal human prostate epithelial cells |
| LPA(2) | inhibition | E2F induction | Phospholipid receptor | [45] | Mouse neuronal cells |
| Dbs | inhibition | E2F induction | Rho-specific guanine nucleotide exchange factor | [45] | Mouse neuronal cells |
| TBX2 | inhibition | TBX2 represses the Cdkn2a (p19(ARF)) promoter | Transcription factor | [31] | Bmi1-/- MEFs |
| TBX3 | inhibition | TBX-3 potently represses expression of both mouse p19(ARF) and human p14(ARF) | Transcription factor | [30] | Mouse neuronal cells |
| Topo1 | promotion | DNA damage-ATM-p53 | Nuclear enzyme regulating DNA structure Relaxes positively and negatively supercoiled DNA | [26] | Normal human cells |
| IGFBP7 | promotion | MEK, ERK pathway In Brafv600E-mediated senescence, IGFBP7 inhibits BRAF-MEK-ERK signaling by inducing RKIP, which prevents BRAF from phosphorylating MEK | Ser/Thr protein kinase, oncogene Growth factor receptor | [25, 46] | Human primary fibroblasts, melanocytes |
| KLF4 | promotion | p53 pathway Suppresses the expression of p53 by directly acting on its promoter Induces p21 | Transcription factor | [47] | Conditionally immortalized MEFs co-expressing RASV12 |
| SAHH | promotion | p53 pathway SAHH inactivation inhibits p53 transcriptional activity | [24b, 28, 48] | Primary human fibroblasts, Primary MEFs | |
| CXCR2 (IL8RB) | promotion | p53 pathway CXCR2 knock-down alleviates both replicative and oncogene-induced senescence and diminishes the DNA-damage response. | Chemokine receptor | [49] | Primary human fibroblasts |
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Collado M , Blasco MA and Serrano M. Cellular senescence in cancer and aging. Cell. 2007; 130: 223 -233. [PubMed] .
- 2. Lowe SW , Cepero E and Evan G. Intrinsic tumour suppression. Nature. 2004; 432: 307 -315. [PubMed] .
- 3. Schmitt CA , Fridman JS , Yang M , Lee S , Baranov E , Hoffman RM and Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002; 109: 335 -346. [PubMed] .
- 4. Xue W , Zender L , Miething C , Dickins RA , Hernando E , Krizhanovsky V , Cordon-Cardo C and Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007; 445: 656 -660. [PubMed] .
- 5. Collado M and Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010; 10: 51 -57. [PubMed] .
- 6. Hahn WC and Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002; 2: 331 -341. [PubMed] .
- 7. Zuckerman V , Wolyniec K , Sionov RV , Haupt S and Haupt Y. Tumour suppression by p53: the importance of apoptosis and cellular senescence. J Pathol. 2009; 219: 3 -15. [PubMed] .
- 8. Parrinello S , Samper E , Krtolica A , Goldstein J , Melov S and Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003; 5: 741 -747. [PubMed] .
- 9. Busuttil RA , Rubio M , Dolle ME , Campisi J and Vijg J. Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell. 2003; 2: 287 -294. [PubMed] .
- 10. Kamijo T , Zindy F , Roussel MF , Quelle DE , Downing JR , Ashmun RA , Grosveld G and Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997; 91: 649 -659. [PubMed] .
- 11. Harvey DM and Levine AJ. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev. 1991; 5: 2375 -2385. [PubMed] .
- 12. Serrano M , Lin AW , McCurrach ME , Beach D and Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88: 593 -602. [PubMed] .
- 13. Kamijo T , Weber JD , Zambetti G , Zindy F , Roussel MF and Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci U S A. 1998; 95: 8292 -8297. [PubMed] .
- 14. Roussel MF p53: regular or super. Cancer Cell. 2002; 2: 434 -435. [PubMed] .
- 15. Luo JL , Yang Q , Tong WM , Hergenhahn M , Wang ZQ and Hollstein M. Knock-in mice with a chimeric human/murine p53 gene develop normally and show wild-type p53 responses to DNA damaging agents: a new biomedical research tool. Oncogene. 2001; 20: 320 -328. [PubMed] .
- 16. Reinbold M , Luo JL , Nedelko T , Jerchow B , Murphy ME , Whibley C , Wei Q and Hollstein M. Common tumour p53 mutations in immortalized cells from Hupki mice heterozygous at codon 72. Oncogene. 2008; 27: 2788 -2794. [PubMed] .
- 17. Song H , Hollstein M and Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007; 9: 573 -580. [PubMed] .
- 18. Dudgeon C , Kek C , Demidov ON , Saito S , Fernandes K , Diot A , Bourdon JC , Lane DP , Appella E , Fornace AJ Jr and Bulavin DV. Tumor susceptibility and apoptosis defect in a mouse strain expressing a human p53 transgene. Cancer Res. 2006; 66: 2928 -2936. [PubMed] .
- 19. Liu Z , Muehlbauer KR , Schmeiser HH , Hergenhahn M , Belharazem D and Hollstein MC. p53 mutations in benzo(a)pyrene-exposed human p53 knock-in murine fibroblasts correlate with p53 mutations in human lung tumors. Cancer Res. 2005; 65: 2583 -2587. [PubMed] .
- 20. Whibley C , Odell AF , Nedelko T , Balaburski G , Murphy M , Liu Z , Stevens L , Walker JH , Routledge M and Hollstein M. Wild-type and HUPKI (human P53 knock-in) murine embryonic fibroblasts: P53/ARF pathway disruption in spontaneous escape from senescence. J Biol Chem. 2010; Epub Feb 4 .
- 21. Quon KC and Berns A. Haplo-insufficiency? Let me count the ways. Genes Dev. 2001; 15: 2917 -2921. [PubMed] .
- 22. Inoue K , Zindy F , Randle DH , Rehg JE and Sherr CJ. Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev. 2001; 15: 2934 -2939. [PubMed] .
- 23. Hannon GJ and Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004; 431: 371 -378. [PubMed] .
- 24. Berns K , Hijmans EM , Mullenders J , Brummelkamp TR , Velds A , Heimerikx M , Kerkhoven RM , Madiredjo M , Nijkamp W , Weigelt B , Agami R , Ge W and Cavet G. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004; 428: 431 -437. [PubMed] .
- 25. Wajapeyee N , Serra RW , Zhu X , Mahalingam M and Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008; 132: 363 -374. [PubMed] .
- 26. Humbert N , Martien S , Augert A , Da Costa M , Mauen S , Abbadie C , de Launoit Y , Gil J and Bernard D. A genetic screen identifies topoisomerase 1 as a regulator of senescence. Cancer Res. 2009; 69: 4101 -4106. [PubMed] .
- 27. Dirac AM and Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem. 2003; 278: 11731 -11734. [PubMed] .
- 28. Leal JF , Fominaya J , Cascon A , Guijarro MV , Blanco-Aparicio C , Lleonart M , Castro ME , Ramon YCS , Robledo M , Beach DH and Carnero A. Cellular senescence bypass screen identifies new putative tumor suppressor genes. Oncogene. 2008; 27: 1961 -1970. [PubMed] .
- 29. Shvarts A , Brummelkamp TR , Scheeren F , Koh E , Daley GQ , Spits H and Bernards R. A senescence rescue screen identifies BCL6 as an inhibitor of anti-proliferative p19(ARF)-p53 signaling. Genes Dev. 2002; 16: 681 -686. [PubMed] .
- 30. Brummelkamp TR , Kortlever RM , Lingbeek M , Trettel F , MacDonald ME , van Lohuizen M and Bernards R. TBX-3, the gene mutated in Ulnar-Mammary Syndrome, is a negative regulator of p19ARF and inhibits senescence. J Biol Chem. 2002; 277: 6567 -6572. [PubMed] .
- 31. Jacobs JJ , Keblusek P , Robanus-Maandag E , Kristel P , Lingbeek M , Nederlof PM , van Welsem T , van de Vijver MJ , Koh EY , Daley GQ and van Lohuizen M. Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat Genet. 2000; 26: 291 -299. [PubMed] .
- 32. Peeper DS , Shvarts A , Brummelkamp T , Douma S , Koh EY , Daley GQ and Bernards R. A functional screen identifies hDRIL1 as an oncogene that rescues RAS-induced senescence. Nat Cell Biol. 2002; 4: 148 -153. [PubMed] .
- 33. Dasari A , Bartholomew JN , Volonte D and Galbiati F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 2006; 66: 10805 -10814. [PubMed] .
- 34. Volonte D , Zhang K , Lisanti MP and Galbiati F. Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Mol Biol Cell. 2002; 13: 2502 -2517. [PubMed] .
- 35. Binet R , Ythier D , Robles AI , Collado M , Larrieu D , Fonti C , Brambilla E , Brambilla C , Serrano M , Harris CC and Pedeux R. WNT16B is a new marker of cellular senescence that regulates p53 activity and the phosphoinositide 3-kinase/AKT pathway. Cancer Res. 2009; 69: 9183 -9191. [PubMed] .
- 36. Kortlever RM , Higgins PJ and Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006; 8: 877 -884. [PubMed] .
- 37. Nogueira V , Park Y , Chen CC , Xu PZ , Chen ML , Tonic I , Unterman T and Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008; 14: 458 -470. [PubMed] .
- 38. Das M , Jiang F , Sluss HK , Zhang C , Shokat KM , Flavell RA and Davis RJ. Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc Natl Acad Sci U S A. 2007; 104: 15759 -15764. [PubMed] .
- 39. MacLaren A , Black EJ , Clark W and Gillespie DA. c-Jun-deficient cells undergo premature senescence as a result of spontaneous DNA damage accumulation. Mol Cell Biol. 2004; 24: 9006 -9018. [PubMed] .
- 40. Utikal J , Polo JM , Stadtfeld M , Maherali N , Kulalert W , Walsh RM , Khalil A , Rheinwald JG and Hochedlinger K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009; 460: 1145 -1148. [PubMed] .
- 41. Banito A , Rashid ST , Acosta JC , Li S , Pereira CF , Geti I , Pinho S , Silva JC , Azuara V , Walsh M , Vallier L and Gil J. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009; 23: 2134 -2139. [PubMed] .
- 42. Gjoerup OV , Wu J , Chandler-Militello D , Williams GL , Zhao J , Schaffhausen B , Jat PS and Roberts TM. Surveillance mechanism linking Bub1 loss to the p53 pathway. Proc Natl Acad Sci U S A. 2007; 104: 8334 -8339. [PubMed] .
- 43. Tarunina M , Alger L , Chu G , Munger K , Gudkov A and Jat PS. Functional genetic screen for genes involved in senescence: role of Tid1, a homologue of the Drosophila tumor suppressor l(2)tid, in senescence and cell survival. Mol Cell Biol. 2004; 24: 10792 -10801. [PubMed] .
- 44. Gil J , Bernard D , Martinez D and Beach D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat Cell Biol. 2004; 6: 67 -72. [PubMed] .
- 45. Kortlever RM , Brummelkamp TR , van Meeteren LA , Moolenaar WH and Bernards R. Suppression of the p53-dependent replicative senescence response by lysophosphatidic acid signaling. Mol Cancer Res. 2008; 6: 1452 -1460. [PubMed] .
- 46. Michaloglou C , Vredeveld LC , Soengas MS , Denoyelle C , Kuilman T , van der Horst CM , Majoor DM , Shay JW , Mooi WJ and Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005; 436: 720 -724. [PubMed] .
- 47. Rowland BD , Bernards R and Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005; 7: 1074 -1082. [PubMed] .
- 48. Leal JF , Ferrer I , Blanco-Aparicio C , Hernandez-Losa J , Ramon YCS , Carnero A and Lleonart ME. S-adenosylhomocysteine hydrolase downregulation contributes to tumorigenesis. Carcinogenesis. 2008; 29: 2089 -2095. [PubMed] .
- 49. Acosta JC , O'Loghlen A , Banito A , Guijarro MV , Augert A , Raguz S , Fumagalli M , Da Costa M , Brown C , Popov N , Takatsu Y , Melamed J and d'Adda di Fagagna F. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008; 133: 1006 -1018. [PubMed] .