



The discovery of Sestrins as p53 targets [1,2] suggested that these proteins are stress-inducible because they protect cells against various insults [2]. Sestrins have dual biochemical functions, as antioxidants that control the activity of peroxiredoxins which scavenge reactive oxygen species (ROS) [3] and as inhibitors of target of rapamycin complex 1 (TORC1) signaling [4,5]. Both ROS accumulation [6] and TORC1 activation [7,8] are associated with accelerated aging and development of age-associated pathologies in diverse organs and organisms, implicating Sestrins as anti-aging agents. Conversely, reduction of ROS accumulation with antioxidants or as a result of TORC1 inhibition [7-12] causes an extension of life span as well as health span. Indeed, we recently confirmed Drosophila Sestrin (dSesn) prevents age-associated pathologies including fat accumulation and cardiac and skeletal muscle degeneration by providing a feedback loop that prevents excessive TORC1 activation and ROS accumulation [4]. In this research perspective, we discuss the possible roles of Sestrins in diverse stress-induced patho-physiological contexts that can result in premature aging and age-related diseases. We suggest that Sestrins provide critical feedback regulation that adjust metabolic and stress responses to different environmental cues and evolutionary constraints.
DNA damage
Chronic exposure to genotoxic stress is known to accelerate aging, and mutations that disrupt proper DNA damage responses and interfere with DNA damage repair are associated with premature aging in humans [13]. Many studies have established that genotoxic stresses can inhibit protein and lipid synthesis, and that these coordinated responses may be essential for survival because reducing energy expenditure on macromolecule biosynthesis can divert scarce resources to the repair of damaged DNA [14]. Sestrins, as DNA damage-inducible proteins, may play a critical role in this process [15]. Both mammalian Sesn1 and Sesn2 are induced upon DNA damage in response to activation of p53 [1,2], and dSesn is also induced upon radiation-induced DNA damage (Figure 1). Increased Sestrin abundance potentiates the activity of AMP-activated protein kinase (AMPK), thereby diminishing TORC1 activity [5]. Reduced TORC1 activity inhibits anabolic pathway including protein and lipid synthesis [16,17]. Shutdown of TORC1-dependent anabolism upon genotoxic stress is likely to be important for minimizing new protein and membrane synthesis and use the energy that was thus saved to promote DNA repair. Therefore, DNA damage-dependent induction of Sestrins may minimize the detrimental effects of DNA damage that contribute to accelerated aging and various pathologies that are associated with premature aging.
Age-dependent accumulation of DNA damage can lead to cancer [13], one of the leading causes of mortality worldwide [18]. Therefore, Sestrin induction in response to DNA damage [1,2] may contribute to the many tumor suppressor functions carried out by p53 [19]. In addition to inhibiting cell proliferation and promoting the death of cells with excessive DNA damage, p53 was recently found to inhibit TORC1 [14] and to suppress cell growth as well as cellular and organismal senescence [19-21]. We found that Sesn1 and/or Sesn2 are critical mediators of p53-induced TORC1 inhibition in cultured cells and in mouse liver [5]. In addition, Sestrins can suppress the growth of some cancer cell lines [2] and loss of Sesn2 makes immortalized cells more susceptible to oncogenic transformation [5]. The SESN1 (6q21) and SESN2 (1p35) loci are frequently deleted in a variety of human cancers [1,22,23], implicating loss of Sestrins in tumor progression and suggesting that Sestrin-dependent inhibition of TORC1 is critical for suppressing tumorigenesis spurred by age-dependent accumulation of damaged DNA.

Figure 1. Induction of dSesn after DNA damage. First instar
fly larvae were challenged with 4000 rads (R) of gamma radiation, and RNA
was extracted after 4 hrs. Northern blot analysis revealed that dSesn
mRNA is highly induced upon irradiation. rp49 mRNA was used as a
loading control.
Oxidative stress
Oxidative stress not only can interfere with the proper flow of genomic information by oxidizing DNA and RNA, but also can damage other macromolecules such as proteins and lipids [6]. Accumulation of oxidative macromolecular damage causes cellular senescence, tissue aging and reduced life span [6], as well as neurodegeneration [24] and metabolic disorders [25], which are diseases associated with aging. Amongst the organelles that are affected by oxidative stress, mitochondria appear to be the most sensitive [6,26]. Moreover, damaged mitochondria are a major source of ROS [6], which escalates oxidative damage in stressed cells. Extensive mitochondrial dysfunction causes cell death, and in some cases can lead to neuronal or muscular degeneration [6,24,27]. To prevent the detrimental consequences of mitochondrial dysfunction, cells eliminate damaged mitochondria through an autophagic process, called mitophagy [28,29].
Sestrins are transcriptionally induced upon oxidative stress [2], and are important for cell survival under oxidative stress [2,3,30,31]. Sestrins can function as oxidoreductases in vitro and in vivo that lead to the reactivation of peroxiredoxin [3], and may be involved in reducing oxidative stress [30-32] by scavenging ROS and/or regenerating reduced peroxiredoxin [3]. Independent of their oxidoreductase activity, Sestrins induce autophagy by inhibition of TORC1 [5,33,34]. Enhanced autophagy results in more efficient elimination of ROS-producing damaged mitochondria in stressed cells [28,29]. Sestrin-induced activation of AMPK and inhibition of TORC1 can also reduce ROS production by increasing the efficiency of mitochondrial respiration [11,12]. Therefore, Sestrins have a key role in maintaining cellular integrity and homeostasis during oxidative insults.
Hypoxia
Hypoxia is another environmental stimulus that can induce Sestrin gene transcription [2]. Sestrins protect cells from apoptosis during hypoxic conditions [2], and Sestrin-induced shutdown of TORC1 signaling can reduce cellular energy consumption that is likely to improve adaptation to hypoxic conditions. Sestrin-stimulated autophagy can provide an additional energy source and at the same time eliminate dysfunctional mitochondria generated by inefficient respiration under low oxygen tension.
Ischemic injury to heart muscles and neurons, which is caused by hypoxia, is one of the major causes of death in elderly individuals [18]. In an experimental model of acute stroke, Sesn2 was shown to be highly induced upon hypoxic injury [2], suggesting that Sesn2 may exert its neuroprotective role during stroke. In the heart, hypoxic injury and re-oxygenation cause bursts of ROS production, which can cause irreversible damage to the heart muscle, resulting in cardiac arrhythmia and heart failure [35,36]. In the Drosophila heart, both aging [37-41] and hypoxia [39,42,43] cause cardiac dysfunction, and activation of TORC1 pathway aggravates or accelerates age-associated arrhythmicity and heart failure [44]. Loss of dSesn function results in a similar cardiac arrhythmicity [4], suggesting a cardio-protective function of Sestrin in restraining TORC1 activity. Thus Sestrin expression retards the appearance of age-associated cardiac pathologies, as was previously observed in response to genetic reduction of TORC1 function [10,44].
Hypoxic preconditioning can protect both heart and neuronal cells from severe ischemic injury-induced cellular damage [36,45], but the underlying mechanisms were not elucidated. Induction of autophagy upon preconditioning was suggested to be required for protection of heart and neuronal cells from hypoxic insults [36,46]. An intriguing possibility to investigate therefore is whether hypoxic preconditioning induces Sestrin to increase the level of autophagy that is required for the prevention of serious heart attacks and neurological strokes.
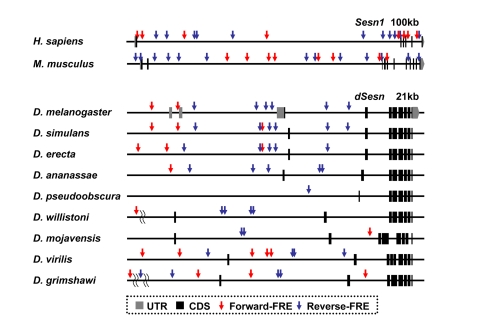
Figure 2. FoxO binding sites in the Sestrin locus vary among the species. Genomic
organization and location of FoxO response elements (FRE, GTAAACAA [57]) in the Sesn1
locus of human and mouse [58] and the dSesn
locus of various Drosophila species [59,60] with indicated
genomic span. Gray boxes indicate untranslated exons (UTR), and black boxes
indicate coding exons (CDS). For Drosophila species other than D.
melanogaster, untranslated regions of dSesn mRNAs are currently
unknown. Forward FREs are indicated by red arrows and reverse FREs by blue
arrows.
An evolvable link to the environment
In addition to the important role that Sestrins play in mediating essential environmental inputs into metabolic regulation, these molecules may also play central roles in responding to other environmental cues such as nutrient supply, hydration status, temperature, chemical damage, and reproductive signals. One might speculate that because these various cues would have different impacts on different organisms, Sestrin genes should have evolved complex cis-regulatory regions to place them under distinct regulatory controls that vary from one organism to another. Indeed, our analysis of Sestrin genomic loci revealed that these sequences are rapidly evolving among the various Drosophila species. For example, the disposition and number of FoxO binding sites in the Sestrin locus vary significantly among species (Figure 2). Comparative studies of cis-regulatory sequences between Drosophila species by swapping these sequences using recombineering techniques may shed light on mechanisms by which selective pressures sculpt the stress response during evolution.
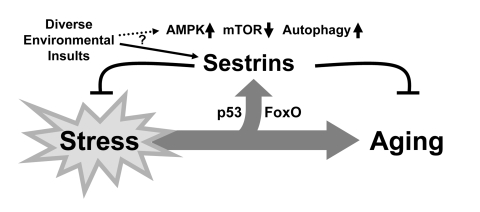
Figure 3. Schematic diagram hypothesizing the role of Sestrin as a brake of stress-accelerated aging processes. Various environmental insults increase
expression of Sestrins to affect Sestrin-mediated regulation of AMPK-TORC1
signaling.
Concluding remarks
In this research perspective, we briefly reviewed how Sestrins may act as suppressors of aging that are responsive to stressful stimuli and insults that can accelerate the aging process. By activating AMPK and inhibiting TORC1, Sestrins can reprogram cells to adapt to stressful conditions by attenuating anabolism and enhancing autophagic catabolism. Sestrins can suppress oxidative damage by acting as either antioxidants, inducers of autophagy that eliminate ROS-producing dysfunctioning mitochondria, or suppressors of ROS-producing mitochondrial metabolism. Therefore, we can view Sestrins as physiological brakes that can attenuate stress-dependent acceleration of aging (Figure 3).
In addition, it is worth noting that Sestrins are also expressed under normal unstressed conditions [1,2,4], and that dSesn knockout mutants show accelerated aging phenotypes even in the absence of any environmental stress [4]. Therefore, it is possible that Sestrins provide a baseline protective function that reduces the damage from physiological insults that are unavoidable consequences of basic life processes such as oxidative respiration and DNA replication.
Over-nutrition and obesity can elevate the incidence and frequency of physiological insults by stimulating TORC1 activity [47], which in turn accelerates anabolic metabolism [16,17]. Hyperactive TORC1 can enhance the accumulation of unfolded and aggregated proteins [48] and ROS [4,12,49], leading to stress responses that increase Sestrin expression, thereby activating negative feedback loops that readjust AMPK and TORC1 activity [4]. Therefore, Sestrins may also function as metabolic brakes that attenuate the pathological consequences of over-nutrition and its associated TORC1 hyperactivity [50]. An interesting human evolutionary question in this regard is whether regulation of Sestrin induction has been weakened in populations subject to frequent starvation conditions since such populations have been shown to be particularly at risk for obesity, presumably as a result of their altered response to nutrient cues [51,52].
Although environmental stress generally accelerates aging, it should be considered that exposure to a low level of stress or stress adaptation can actually be beneficial for the organism, increasing life span and preventing age-associated degenerative diseases [53-56]. The beneficial effect of low-level stress exposure, referred to as hormesis, was observed in both experimental animal models and human subjects [53-56], but no decisive molecular explanation has been provided to explain this paradoxical phenomenon. Given that Sestrins are upregulated in response to a variety of stresses [1,2], it will be interesting to investigate whether stress-induced Sestrins are also mediators of the hormetic effect.
In summary, we suggest that Sestrins are uniquely poised at the crossroads between stress and aging to adjust the metabolic timbre of an organism to meet its needs under normal conditions and to respond to predictable forms of environmental stress. Future experiments should shed light on the specific mechanisms by which the various effector functions of the Sestrins are achieved.
Acknowledgments
The authors thank Charles L. Sanders and Myungjin Kim for their encouragement and intuitive suggestions. Work was supported by grants from the National Institutes of Health and the Superfund Research Program (CA118165, ES006376 and P42-ES010337 to M.K., and NS29870 and AI070654 to E.B.), National Science Foundation (IOS-0744662 to E.B.), Korea Research Foundation (KRF-2007-357-C00096 to J.H.L.), and Human Frontier Science Program Organzation (LT00653/2008-L to J.H.L). M.K. is an American Cancer Society Professor.
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Velasco-Miguel S , Buckbinder L , Jean P , Gelbert L , Talbott R , Laidlaw J , Seizinger B and Kley N. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene. 1999; 18: 127 -137. [PubMed] .
- 2. Budanov AV , Shoshani T , Faerman A , Zelin E , Kamer I , Kalinski H , Gorodin S , Fishman A , Chajut A , Einat P , Skaliter R , Gudkov AV and Chumakov PM. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002; 21: 6017 -6031. [PubMed] .
- 3. Budanov AV , Sablina AA , Feinstein E , Koonin EV and Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004; 304: 596 -600. [PubMed] .
- 4. Lee JH , Budanov AV , Park EJ , Birse R , Kim TE , Perkins GA , Ocorr K , Ellisman MH , Bodmer R , Bier E and Karin M. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010; 327: 1223 -1228. [PubMed] .
- 5. Budanov AV and Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008; 134: 451 -460. [PubMed] .
- 6. Finkel T and Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000; 408: 239 -247. [PubMed] .
- 7. Stanfel MN , Shamieh LS , Kaeberlein M and Kennedy BK. The TOR pathway comes of age. Biochim Biophys Acta. 2009; 1790: 1067 -1074. [PubMed] .
- 8. Kapahi P and Zid B. TOR pathway: linking nutrient sensing to life span. Sci Aging Knowledge Environ. 2004; 2004: PE34 [PubMed] .
- 9. Landis GN and Tower J. Superoxide dismutase evolution and life span regulation. Mech Ageing Dev. 2005; 126: 365 -379. [PubMed] .
- 10. Luong N , Davies CR , Wessells RJ , Graham SM , King MT , Veech R , Bodmer R and Oldham SM. Activated FOXO-mediated insulin resistance is blocked by reduction of TOR activity. Cell Metab. 2006; 4: 133 -142. [PubMed] .
- 11. Zid BM , Rogers A , Katewa SD , Au Lu T , Benzer S and Kapahi P. 4E-BP modulates lifespan and mitochondrial translation upon dietary restriction in Drosophila. Cell. 2009; 139: 149 -160. [PubMed] .
- 12. Bonawitz ND , Chatenay-Lapointe M , Pan Y and Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007; 5: 265 -277. [PubMed] .
- 13. Hoeijmakers JH DNA damage, aging, and cancer. N Engl J Med. 2009; 361: 1475 -1485. [PubMed] .
- 14. Levine AJ , Feng Z , Mak TW , You H and Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006; 20: 267 -275. [PubMed] .
- 15. Hay N p53 strikes mTORC1 by employing sestrins. Cell Metab. 2008; 8: 184 -185. [PubMed] .
- 16. Hay N and Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004; 18: 1926 -1945. [PubMed] .
- 17. Laplante M and Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009; 19: R1046 -1052. [PubMed] .
- 18. World Health Organization Geneva World Health Organization The world health report 2008: primary health care now more than ever. 2008; .
- 19. Vousden KH and Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009; 137: 413 -431. [PubMed] .
- 20. Demidenko ZN , Korotchkina LG , Gudkov AV and Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660 -9664. [PubMed] .
- 21. Matheu A , Maraver A , Klatt P , Flores I , Garcia-Cao I , Borras C , Flores JM , Vina J , Blasco MA and Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007; 448: 375 -379. [PubMed] .
- 22. Schwab M , Praml C and Amler LC. Genomic instability in 1p and human malignancies. Genes Chromosomes Cancer. 1996; 16: 211 -229. [PubMed] .
- 23. Ragnarsson G , Eiriksdottir G , Johannsdottir JT , Jonasson JG , Egilsson V and Ingvarsson S. Loss of heterozygosity at chromosome 1p in different solid human tumours: association with survival. Br J Cancer. 1999; 79: 1468 -1474. [PubMed] .
- 24. Emerit J , Edeas M and Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004; 58: 39 -46. [PubMed] .
- 25. Roberts CK and Sindhu KK. Oxidative stress and metabolic syndrome. Life Sci. 2009; 84: 705 -712. [PubMed] .
- 26. Hyslop PA , Hinshaw DB , Halsey WA Jr , Schraufstatter IU , Sauerheber RD , Spragg RG , Jackson JH and Cochrane CG. Mechanisms of oxidant-mediated cell injury. The glycolytic and mitochondrial pathways of ADP phosphorylation are major intracellular targets inactivated by hydrogen peroxide. J Biol Chem. 1988; 263: 1665 -1675. [PubMed] .
- 27. Green DR and Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004; 305: 626 -629. [PubMed] .
- 28. Jin S Autophagy, mitochondrial quality control, and oncogenesis. Autophagy. 2006; 2: 80 -84. [PubMed] .
- 29. Yen WL and Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda). 2008; 23: 248 -262. [PubMed] .
- 30. Papadia S , Soriano FX , Leveille F , Martel MA , Dakin KA , Hansen HH , Kaindl A , Sifringer M , Fowler J , Stefovska V , McKenzie G , Craigon M and Corriveau R. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008; 11: 476 -487. [PubMed] .
- 31. Lipton SA NMDA receptor activity regulates transcription of antioxidant pathways. Nat Neurosci. 2008; 11: 381 -382. [PubMed] .
- 32. Nogueira V , Park Y , Chen CC , Xu PZ , Chen ML , Tonic I , Unterman T and Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008; 14: 458 -470. [PubMed] .
- 33. Maiuri MC , Malik SA , Morselli E , Kepp O , Criollo A , Mouchel PL , Carnuccio R and Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle. 2009; 8: 1571 -1576. [PubMed] .
- 34. Chan EY mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009; 2: pe51 [PubMed] .
- 35. Giordano FJ Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005; 115: 500 -508. [PubMed] .
- 36. Gottlieb RA and Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010; 72: 45 -59. [PubMed] .
- 37. Taghli-Lamallem O , Akasaka T , Hogg G , Nudel U , Yaffe D , Chamberlain JS , Ocorr K and Bodmer R. Dystrophin deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell. 2008; 7: 237 -249. [PubMed] .
- 38. Wessells RJ , Fitzgerald E , Cypser JR , Tatar M and Bodmer R. Insulin regulation of heart function in aging fruit flies. Nat Genet. 2004; 36: 1275 -1281. [PubMed] .
- 39. Ocorr K , Akasaka T and Bodmer R. Age-related cardiac disease model of Drosophila. Mech Ageing Dev. 2007; 128: 112 -116. [PubMed] .
- 40. Ocorr K , Reeves NL , Wessells RJ , Fink M , Chen HS , Akasaka T , Yasuda S , Metzger JM , Giles W , Posakony JW and Bodmer R. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc Natl Acad Sci U S A. 2007; 104: 3943 -3948. [PubMed] .
- 41. Cammarato A , Dambacher CM , Knowles AF , Kronert WA , Bodmer R , Ocorr K and Bernstein SI. Myosin transducer mutations differentially affect motor function, myofibril structure, and the performance of skeletal and cardiac muscles. Mol Biol Cell. 2008; 19: 553 -562. [PubMed] .
- 42. Feala JD , Omens JH , Paternostro G and McCulloch AD. Discovering regulators of the Drosophila cardiac hypoxia response using automated phenotyping technology. Ann N Y Acad Sci. 2008; 1123: 169 -177. [PubMed] .
- 43. Akasaka T , Klinedinst S , Ocorr K , Bustamante EL , Kim SK and Bodmer R. The ATP-sensitive potassium (KATP) channel-encoded dSUR gene is required for Drosophila heart function and is regulated by tinman. Proc Natl Acad Sci U S A. 2006; 103: 11999 -12004. [PubMed] .
- 44. Wessells R , Fitzgerald E , Piazza N , Ocorr K , Morley S , Davies C , Lim HY , Elmen L , Hayes M , Oldham S and Bodmer R. d4eBP acts downstream of both dTOR and dFoxo to modulate cardiac functional aging in Drosophila. Aging Cell. 2009; 8: 542 -552. [PubMed] .
- 45. Gidday JM , Fitzgibbons JC , Shah AR and Park TS. Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci Lett. 1994; 168: 221 -224. [PubMed] .
- 46. Park HK , Chu K , Jung KH , Lee ST , Bahn JJ , Kim M , Lee SK and Roh JK. Autophagy is involved in the ischemic preconditioning. Neurosci Lett. 2009; 451: 16 -19. [PubMed] .
- 47. Dann SG , Selvaraj A and Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007; 13: 252 -259. [PubMed] .
- 48. Ozcan U , Ozcan L , Yilmaz E , Duvel K , Sahin M , Manning BD and Hotamisligil GS. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008; 29: 541 -551. [PubMed] .
- 49. Kim JH , Chu SC , Gramlich JL , Pride YB , Babendreier E , Chauhan D , Salgia R , Podar K , Griffin JD and Sattler M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005; 105: 1717 -1723. [PubMed] .
- 50. Topisirovic I and Sonenberg N. Cell biology. Burn out or fade away. Science. 2010; 327: 1210 -1211. [PubMed] .
- 51. Carulli L , Rondinella S , Lombardini S , Canedi I , Loria P and Carulli N. Review article: diabetes, genetics and ethnicity. Aliment Pharmacol Ther. 2005; 22 Suppl 2: 16 -19. [PubMed] .
- 52. Neel JV Diabetes mellitus: a "thrifty" genotype rendered detrimental by "progress". Am J Hum Genet. 1962; 14: 353 -362. [PubMed] .
- 53. Mattson MP and Calabrese EJ. New York Humana Press (eds.) Hormesis: A Revolution in Biology, Toxicology and Medicine. 2009; .
- 54. Sanders CL New York Springer Radiation Hormesis and the Linear-No-Threshold Assumption. 2010; .
- 55. Le Bourg E Hormesis, aging and longevity. Biochim Biophys Acta. 2009; 1790: 1030 -1039. [PubMed] .
- 56. Minois N Longevity and aging: beneficial effects of exposure to mild stress. Biogerontology. 2000; 1: 15 -29. [PubMed] .
- 57. Furuyama T , Nakazawa T , Nakano I and Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J. 2000; 349: 629 -634. [PubMed] .
- 58. Hubbard TJ , Aken BL , Ayling S , Ballester B , Beal K , Bragin E , Brent S , Chen Y , Clapham P , Clarke L , Coates G , Fairley S and Fitzgerald S. Ensembl 2009. Nucleic Acids Res. 2009; 37: D690 -697. [PubMed] .
- 59. Clark AG , Eisen MB , Smith DR , Bergman CM , Oliver B , Markow TA , Kaufman TC , Kellis M , Gelbart W , Iyer VN , Pollard DA , Sackton TB and Larracuente AM. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007; 450: 203 -218. [PubMed] .
- 60. Stark A , Lin MF , Kheradpour P , Pedersen JS , Parts L , Carlson JW , Crosby MA , Rasmussen MD , Roy S , Deoras AN , Ruby JG , Brennecke J and Hodges E. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007; 450: 219 -232. [PubMed] .