SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence
Abstract
Members of the signal transducers and activators of transcription (STATs) family of proteins, which connect cytokine signaling to activation of transcription, are frequently activated in human cancers. Suppressors of cytokine signaling (SOCS) are transcriptional targets of activated STAT proteins that negatively control STAT signaling. SOCS1 expression is silenced in multiple human cancers suggesting a tumor suppressor role for this protein. However, SOCS1 not only regulates STAT signaling but can also localize to the nucleus and directly interact with the p53 tumor suppressor through its central SH2 domain. Furthermore, SOCS1 contributes to p53 activation and phosphorylation on serine 15 by forming a ternary complex with ATM or ATR. Through this mechanism SOCS1 regulates the process of oncogene-induced senescence, which is a very important tumor suppressor response. A mutant SOCS1 lacking the SOCS box cannot interact with ATM/ATR, stimulate p53 or induce the senescence phenotype, suggesting that the SOCS box recruits DNA damage activated kinases to its interaction partners bound to its SH2 domain. Proteomic analysis of SOCS1 interaction partners revealed other potential targets of SOCS1 in the DNA damage response. These newly discovered functions of SOCS1 help to explain the increased susceptibility of Socs1 null mice to develop cancer as well as their propensity to develop autoimmune diseases. Consistently, we found that mice lacking SOCS1 displayed defects in the regulation of p53 target genes including Mdm2, Pmp22, PUMA and Gadd45a. The involvement of SOCS1 in p53 activation and the DNA damage response defines a novel tumor suppressor pathway and intervention point for future cancer therapeutics.
SOCS1, cancer and senescence
Cytokines are secreted proteins that regulate
different cellular processes including survival, proliferation and
differentiation. Following binding to their receptors, cytokines activate the
Janus kinases (JAK1, JAK2, JAK3 and Tyk2) leading to the phosphorylation of
tyrosine residues on the cytoplasmic portion of the receptor creating docking
sites for signaling molecules containing a SH2 domain [1,2]. Members of the
STAT
family of proteins that are recruited to the phosphorylated
cytokine receptors themselves become phosphorylation substrates for JAK
kinases. Phosphorylated STAT proteins homo- or hetero- dimerize and translocate
to the nucleus to activate transcription of target genes by binding to specific
response elements in their promoter regions. Among these cytokine-induced
proteins, members of the SOCS family constitute important negative regulators
of the JAK/STAT signaling pathway.
There are eight members of the SOCS family of proteins
(CIS, SOCS1-7), each of which harbor a central SH2 domain and a C-terminal SOCS
box region [3] (Figure 1). The suppressor of cytokine signaling SOCS1 was
initially identified as a cytokine-inducible inhibitor of STAT signaling
[4,5,6]. Through its SH2 domain, SOCS1 can directly bind phosphorylated JAK2
to prevent the phosphorylation of STAT. SOCS1 also possesses a kinase
inhibitory region (KIR), a domain composed of less than 30 amino acids, which
shares homology with the pseudosubstrate inhibitory region of JAK and leads to inhibition
of the catalytic activity of JAK [7,8]. The SOCS box allows recruitment of
elongin B/C and Cullin 2 to form an ubiquitin E3 ligase complex [9,10]. This
allows the SOCS protein to operate as an adaptor to trigger ubiquitination and
degradation of proteins involved in cellular signaling including JAK [11],
TEL-JAK2 [12], IRS-1/2 [13], FAK [14], Vav [15] and Mal [16]. It is currently
thought that SOCS1 contributes to tumor suppression due to its ability to
control and terminate the activation of STATs [17,18,19,20,21,22,23,24,25]. On
the other hand, the relationship between SOCS1 and other tumor suppressor pathways and the cellular mechanisms by
which SOCS1 might exert its tumor suppression remain largely unexplored.
To prevent the formation of cancer, normal cells
possess intrinsic tumor suppressor mechanisms that are triggered upon oncogene
activation. Like apoptosis, cellular senescence opposes cellular transformation
by limiting the proliferation of cells expressing oncogenes. In normal human
diploid cells, oncogene activation causes a permanent growth arrest with
features of cellular senescence [26]. We have recently extended the list of
oncogenes known to trigger the senescence response to include the JAK/STAT5
pathway. The transcription factor STAT5 is implicated in tumor formation by
regulating important cellular processes including cell cycle progression,
apoptosis, angiogenesis and metastasis [27]. However, in normal cells,
expression of Tel/Jak2 or constitutively activated allele of STAT5A and B initiated
a cell cycle arrest in G1 associated with markers of premature cellular
senescence and activation of the tumor suppressors Rb and p53 [28,29,30].
SOCS box proteins and the regulation of p53
The activation of the p53 pathway following oncogene
activation is crucial to induce senescence in normal cells. In mice,
stimulation of p53 is dependent on p19ARF (Alternative Reading Frame), which is
induced by several oncogenes [31,32]. However, the role of ARF in oncogene-induced
senescence in human cells is still unclear [33]. In order to identify new
regulators of p53 activation following constitutively activated STAT5
expression in normal cells, we performed microarray analysis covering the
entire human transcriptome. We observed that the expression of SOCS1 was highly
increased at both mRNA and protein level during STAT5-induced senescence [34].
Unexpectedly, SOCS1 expression in normal human fibroblasts was sufficient to
trigger a p53-dependent cell cycle arrest displaying features of the senescence
phenotype. This function of SOCS1 was dependent on the integrity of its SOCS
box. In addition, SOCS1, but not a mutant lacking the SOCS box domain, led to
the accumulation of phosphorylated p53 on serine 15 and increased transcription
of the p53 target gene p21CIP. The knockdown of SOCS1 during STAT5-induced
senescence reduced the phosphorylation of p53 on Ser15, diminished the nuclear
accumulation of p53 and compromised the development of senescence phenotype
[34]. The remaining activated p53 and partial bypass of the senescence response
observed following the knockdown of SOCS1 might arise from the ability of STAT5
to engage multiple signaling pathways to ensure p53 activation. For example,
STAT5 can directly transactivate the promoter of the PML gene and stimulate its
expression in a p53-independent fashion [30]. The PML protein can then inhibit
Mdm2 and stimulate p53 [35,36] contributing to the senescence phenotype
[37,38].
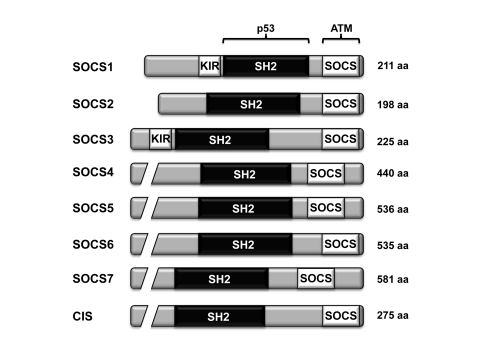
Figure 1. The domain architecture of the different members of the SOCS family of proteins. All eight members of the SOCS family
harbor a central SH2 domain and a C-terminal SOCS box. Both SOCS1 and
SOCS3 also contain a kinase inhibitory region (KIR). The region of
SOCS1 interacting with p53 and ATM are shown [34].
SOCS1 mediated STAT5-induced senescence via an unexpected
protein-protein interaction between the SH2 domain of SOCS1 and the
transactivation domain of p53 [34]. Because the transactivation domain of p53
harbors no tyrosine residues, the binding should occur independently of
tyrosine phosphorylation, as reported before for SOCS1 binding to Vav [15] and
for other SH2 domains as well [39,40]. The von Hippel-Lindau protein (VHL),
another SOCS box-containing protein, has been recently shown to interact with
p53. This interaction does not rely on an SH2 domain but on the SOCS box domain
of VHL. However, like SOCS1, VHL facilitates p53 interaction with the DNA
damage activated kinase ATM [41]. Hence, SOCS1 links DNA damage signals
stimulated by oncogenic activity to p53.
Interestingly, SOCS1 is not the
only protein inhibitor of STAT implicated in the regulation of p53 activity.
The protein inhibitors of activated STAT, PIAS1 and PIASy both promote the
sumoylation and transcriptional activity of p53 [42,43,44]. However, the
mechanism of activation of p53 by PIAS is still unclear. While the sumoylation
of p53 by PIAS1 has been demonstrated [43], a mutated PIAS1 lacking the RING
finger-like domain and defective in promoting p53 sumoylation was sufficient to
activate p53 [44]. Furthermore, by controlling the activity of both p53 and Rb,
PIASy regulates Ras-induced senescence and apoptosis [42]. These data suggest
that the control of STAT signaling is tightly linked to the activation of p53
to possibly control the JAK/STAT oncogenic pathway.
Inhibitors of STATs activity and the DNA damage
response
The stimulation of p53 during oncogene-induced
senescence is associated with the activation of the DNA damage response
[28,45,46]. The DNA damage observed in normal cells expressing activated
oncogenes may be due to reactive oxygen species [47] and/or some type of
replicative stress [45,46]. SOCS1-induced senescence was accompanied by the
activation of the DNA damage-regulated kinases ATM and Chk2. Since the
stimulation of p53 reporters by SOCS1 was partially blocked in cells depleted
of ATM, ATM might participate in the SOCS1-dependent activation of p53. Using
pulldown assays, we demonstrated that SOCS1 interacted with both ATM and ATR
through its SOCS box (Figure 1) [34]. ATM is an important mediator of the
senescence response by activating the p53 pathway, mainly through
phosphorylation of the Ser 15 residue [28,45,46]. Depletion of SOCS1 during
STAT5-induced senescence caused a dramatic decrease in Ser15 phosphorylation of
p53. In order to form a ternary complex with p53 and ATM, SOCS1 must localize
to the nucleus. We confirmed that SOCS1 is able to localize to the nucleus and
that endogenous SOCS1 colocalized to DNA damage foci with ATM during
STAT5-induced senescence [34], thus reinforcing the notion that SOCS1 is a
mediator of the DNA damage response. Not only SOCS1 but also other proteins
controlling JAK/STAT signaling are known to localize to DNA damage sites. PIAS1
and PIAS4 were also shown to localize to DNA breaks and contribute to the DNA
damage response by sumoylating BRCA1 [48,49]. Together, these findings
strongly suggest a close link between cytokine signaling and the DNA damage
response.
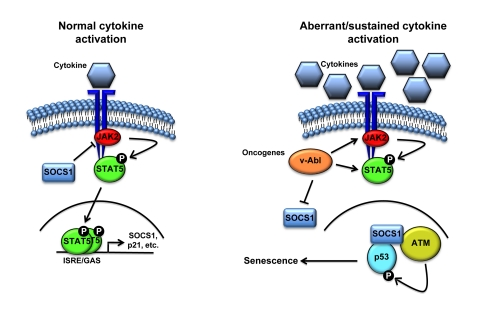
Figure 2. Schematic representation of the cell proliferation control exerted by SOCS1. Following
activation of the receptor by cytokine binding, JAK phosphorylates the
receptor creating a docking site for STATs. JAK then phopshorylates STATs
causing its release from the receptor, allowing dimerization and
translocation to the nucleus to activate the transcription of specific
genes including members of the SOCS family. Subsequently, SOCS terminates
cytokine signaling by blocking JAK activity and STAT recruitment to the
receptor. However, aberrant activation of STAT5 triggered by oncogenic
fusion kinases like TEL-JAK2 might result in sustained levels of SOCS1 that
can activate p53 by forming a complex with ATM and p53.
Cytokines, senescence and SOCS1: an emergency switch
to control proliferation
Senescent cells secrete numerous
cytokines and other mediators that modify the tissue microenvironment. The sum
of these secreted factors constitutes what has been named the
senescence-associated secretory phenotype (SASP) [50]. Among the SASP factors,
IL-6 is required for the oncogene-induced senescence and induction of the tumor
suppressor p15INK4B [51]. Furthermore, persistent, but not transient, DNA
damage signaling triggers the ATM-dependent IL-6 secretion, presumably to call
attention to the presence of damaged cells [52]. During oncogene-induced senescence,
IL-6 also amplifies the secretion of IL-8 [51], which with GROαactivates the CXCR2 receptor to
reinforce senescence [53]. Among the factors secreted by senescent cells,
IGFBP7 [54] and PAI-1 [55] contribute to the growth arrest response, while p53
regulates expression of chemokines directing the immune system to permit the
clearance of senescent cells [56]. Collectively, these reports suggest that
cytokine signaling could prevent tumor formation by promoting cellular
senescence.
The capacity of SOCS1 to activate the p53 pathway can
establish an emergency anti-proliferative program in cells exposed to sustain
or aberrant cytokine stimulation (Figure 2). Following normal activation of the
JAK/STAT pathway, SOCS1 blocks the phosphorylation of STAT by inhibiting or
degrading JAK2. However, aberrant and sustained stimulation of STAT might
induce a molecular switch allowing SOCS1 to localize to DNA breaks and
stimulate ATM-dependent activation of p53.
A general role for SOCS1 in the DNA damage response
The localization of SOCS1 to DNA breaks during
STAT5-induced senescence raises numerous questions. First, does the SOCS1
ubiquitin ligase activity contribute to the DNA damage response? A novel
cascade of ubiquitination controlled by the E3 ubiquitin ligases RNF8/RNF168
and HERC2 have recently been reported to control the recruitment of BRCA1 and
53BP1 by ubiquitinating the histones H2A and H2AX [57,58,59,60,61,62]. The
presence of SOCS1 at DNA breaks could not only regulate ATM-mediated p53
activation but also control the DNA repair process. Second, what are the
mechanisms underlying the nuclear transport of SOCS1 and its presence at DNA
damage foci? Since most of its interacting partners were localized to the
plasma membrane, SOCS1 was considered to be mostly a cytoplasmic protein, but
recent evidences suggest that it can localize to the nucleus under certain
conditions including STAT5-induced senescence [34,63]. A bipartite nuclear
localization signal (NLS) located between the SH2 domain and the SOCS box
allows nuclear localization of SOCS1 [63,64]. However, the mechanism
controlling the active transport of SOCS1 remains unclear. A clearer
understanding of the mechanisms controlling SOCS1 nuclear localization would be
crucial to determine how SOCS1 mediates its tumor suppressor activity.
Post-translational modifications like ubiquitination and phosphorylation that
have been shown to control the nuclear localization of p53 [65,66,67] and STAT
[68] could also control the nucleo-cytoplasmic shuttling of SOCS1. Exclusion of
SOCS1 from the nucleus would prevent the formation of the ternary complex with p53
and ATM, preventing the activation of p53. Furthermore, the phosphorylation
status of SOCS1 could regulate its activity since aberrant SOCS1
phosphorylation is associated with cellular transformation. Actually, phosphorylation
of SOCS1 triggered by the oncogenic v-Abl kinase impedes the SOCS1-Elongin B/C
interaction, leading to sustained JAK/STAT signaling [69]. v-Abl signaling
induces multiple serine/threonine kinases including members of the Pim kinase
family. Pim-1 and Pim-2 are required for efficient cellular transformation
mediated by v-Abl [70] and are able to phosphorylate SOCS1 and disrupt its
binding to Elongin C [71]. Because SOCS1 requires the SOCS box to form a
complex with ATM, v-Abl- or Pim kinase-mediated phosphorylation could
potentially interfere with this interaction and block p53 activation.
Therefore, it appears that aberrant phosphorylation by oncogenic kinases could
interfere with the tumor suppressor activities of SOCS1 by at least two
different mechanisms: phosphorylated SOCS1 would not be able to inhibit the
JAK/STAT pathway and to interact with ATM and promote p53 activation.
Table I. Identification of SOCS1 interaction partners by mass spectrometry*.
| Protein
| Function |
|
Elongin
C
|
Interacts
with SOCS box [10]
|
|
Elongin
B
|
Interacts
with SOCS box [10]
|
|
Pericentrin
|
Cells
depleted of pericentrin enter senescence due to p53 activation [72].
|
|
SHC
(Src homology 2 domain containing) transforming protein 1 (SHC1)
|
Member
of the Shc protein family of molecular adaptors, SHC1 promotes apoptosis by
its redox activity. SHC1 is implicated in the control of oxidative stress and
life span in mammals [73].
|
|
Tripartite
motif-containing 28 (TRIM28 or KAP1)
|
TRIM28
is implicated in transcriptional control through its interaction with the
Kruppel-associated box repression domain. TRIM28 contributes to DNA repair
mechanisms [74].
|
|
5'-nucleotidase,
cytosolic II (NT5C2)
|
NT5C2
hydrolyzes 5-prime-monophosphate (IMP) and other purine nucleotides. NT5C2 is
implicated in the maintenance of a constant composition of intracellular
purine/pyrimidine nucleotides [75].
|
|
BCL2-associated
transcription factor 1 (BCLAF1)
|
BCLAF1,
a transcriptional repressor that interacts with members of the BCL2 family of
proteins, promotes apoptosis [76].
|
|
Human
positive cofactor 4 (PC4)
|
Suppressor
of oxidative mutator phenotype [77].
Accumulates at DNA damage foci [78].
|
Finally, the role of SOCS1 as a
mediator facilitating the interactions of ATM and ATR with their targets
suggests that other interaction partners of SOCS1 could also become the
substrates of ATM/ATR-dependent phosphorylation during the DNA damage
response. Proteomic analysis of SOCS1 complexes revealed putative interactions
with several proteins that play a role in the DNA damage response, apoptosis or
oxidative stress pathways (Table I). Future work will determine which functions
of SOCS1 apply to every one of its interaction partners: ubiquitination
followed by proteolytic degradation or DNA damage stimulated phosphorylation.
Conclusions
Studies on molecular mechanisms underlying cellular
senescence have made significant contributions to the discovery of novel
regulators of tumor suppressor pathways. Using microarrays or cDNA / siRNA
screens, multiple researchers have identified novel regulators of p53 or Rb in
controlling tumor formation. Using this approach to study STAT5-induced
senescence, we identified SOCS1 as an important activator of the p53 and the
DNA damage response. Surprisingly, the SOCS box represents a binding motif for
ATM and ATR [34]. To date, about 40 proteins are known to harbor a SOCS box
domain. Clearly further work will determine whether SOCS box-containing
proteins also participate in the DNA damage response and control oncogenesis.
Acknowledgments
We thank Gillian Vogel for critical reading of the
manuscript and helpful suggestions. F.A.M. and G.F. are supported by the Fonds
de Recherche en Santé du Québec and V.C. by the Natural Sciences and
Engineering Research Council of Canada (NSERC). This work was funded by grants
from the Canadian Institutes of Health Research (CIHR MOP82887 to G.F. and
MOP84234 to S.I.).
Conflicts of Interest
The authors of this manuscript have no conflict of
interests to declare.
References
-
1.
Hanada
T
and Yoshimura
A.
Regulation of cytokine signaling and inflammation.
Cytokine Growth Factor Rev.
2002;
13:
413
-421.
[PubMed]
.
-
2.
Ward
AC
, Touw
I
and Yoshimura
A.
The Jak-Stat pathway in normal and perturbed hematopoiesis.
Blood.
2000;
95:
19
-29.
[PubMed]
.
-
3.
Alexander
WS
Suppressors of cytokine signalling (SOCS) in the immune system.
Nat Rev Immunol.
2002;
2:
410
-416.
[PubMed]
.
-
4.
Endo
TA
, Masuhara
M
, Yokouchi
M
, Suzuki
R
, Sakamoto
H
, Mitsui
K
, Matsumoto
A
, Tanimura
S
, Ohtsubo
M
, Misawa
H
, Miyazaki
T
, Leonor
N
and Taniguchi
T.
A new protein containing an SH2 domain that inhibits JAK kinases.
Nature.
1997;
387:
921
-924.
[PubMed]
.
-
5.
Naka
T
, Narazaki
M
, Hirata
M
, Matsumoto
T
, Minamoto
S
, Aono
A
, Nishimoto
N
, Kajita
T
, Taga
T
, Yoshizaki
K
, Akira
S
and Kishimoto
T.
Structure and function of a new STAT-induced STAT inhibitor.
Nature.
1997;
387:
924
-929.
[PubMed]
.
-
6.
Starr
R
, Willson
TA
, Viney
EM
, Murray
LJ
, Rayner
JR
, Jenkins
BJ
, Gonda
TJ
, Alexander
WS
, Metcalf
D
, Nicola
NA
and Hilton
DJ.
A family of cytokine-inducible inhibitors of signalling.
Nature.
1997;
387:
917
-921.
[PubMed]
.
-
7.
Nicholson
SE
, Willson
TA
, Farley
A
, Starr
R
, Zhang
JG
, Baca
M
, Alexander
WS
, Metcalf
D
, Hilton
DJ
and Nicola
NA.
Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction.
EMBO J.
1999;
18:
375
-385.
[PubMed]
.
-
8.
Yasukawa
H
, Misawa
H
, Sakamoto
H
, Masuhara
M
, Sasaki
A
, Wakioka
T
, Ohtsuka
S
, Imaizumi
T
, Matsuda
T
, Ihle
JN
and Yoshimura
A.
The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop.
EMBO J.
1999;
18:
1309
-1320.
[PubMed]
.
-
9.
Kamura
T
, Maenaka
K
, Kotoshiba
S
, Matsumoto
M
, Kohda
D
, Conaway
RC
, Conaway
JW
and Nakayama
KI.
VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases.
Genes Dev.
2004;
18:
3055
-3065.
[PubMed]
.
-
10.
Kamura
T
, Sato
S
, Haque
D
, Liu
L
, Kaelin
WG Jr
, Conaway
RC
and Conaway
JW.
The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families.
Genes Dev.
1998;
12:
3872
-3881.
[PubMed]
.
-
11.
Ungureanu
D
, Saharinen
P
, Junttila
I
, Hilton
DJ
and Silvennoinen
O.
Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1.
Mol Cell Biol.
2002;
22:
3316
-3326.
[PubMed]
.
-
12.
Kamizono
S
, Hanada
T
, Yasukawa
H
, Minoguchi
S
, Kato
R
, Minoguchi
M
, Hattori
K
, Hatakeyama
S
, Yada
M
, Morita
S
, Kitamura
T
, Kato
H
and Nakayama
K.
The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2.
J Biol Chem.
2001;
276:
12530
-12538.
[PubMed]
.
-
13.
Rui
L
, Yuan
M
, Frantz
D
, Shoelson
S
and White
MF.
SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2.
J Biol Chem.
2002;
277:
42394
-42398.
[PubMed]
.
-
14.
Liu
E
, Cote
JF
and Vuori
K.
Negative regulation of FAK signaling by SOCS proteins.
EMBO J.
2003;
22:
5036
-5046.
[PubMed]
.
-
15.
De
Sepulveda P
, Ilangumaran
S
and Rottapel
R.
Suppressor of cytokine signaling-1 inhibits VAV function through protein degradation.
J Biol Chem.
2000;
275:
14005
-14008.
[PubMed]
.
-
16.
Mansell
A
, Smith
R
, Doyle
SL
, Gray
P
, Fenner
JE
, Crack
PJ
, Nicholson
SE
, Hilton
DJ
, O'Neill
LA
and Hertzog
PJ.
Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation.
Nat Immunol.
2006;
7:
148
-155.
[PubMed]
.
-
17.
Rottapel
R
, Ilangumaran
S
, Neale
C
, La Rose
J
, Ho
JM
, Nguyen
MH
, Barber
D
, Dubreuil
P
and de Sepulveda
P.
The tumor suppressor activity of SOCS-1.
Oncogene.
2002;
21:
4351
-4362.
[PubMed]
.
-
18.
Fukushima
N
, Sato
N
, Sahin
F
, Su
GH
, Hruban
RH
and Goggins
M.
Aberrant methylation of suppressor of cytokine signalling-1 (SOCS-1) gene in pancreatic ductal neoplasms.
Br J Cancer.
2003;
89:
338
-343.
[PubMed]
.
-
19.
Galm
O
, Yoshikawa
H
, Esteller
M
, Osieka
R
and Herman
JG.
SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma.
Blood.
2003;
101:
2784
-2788.
[PubMed]
.
-
20.
Jiang
S
, Zhang
HW
, Lu
MH
, He
XH
, Li
Y
, Gu
H
, Liu
MF
and Wang
ED.
MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene.
Cancer Res.
2010;
70:
3119
-3127.
[PubMed]
.
-
21.
Melzner
I
, Bucur
AJ
, Bruderlein
S
, Dorsch
K
, Hasel
C
, Barth
TF
, Leithauser
F
and Moller
P.
Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line.
Blood.
2005;
105:
2535
-2542.
[PubMed]
.
-
22.
Pichiorri
F
, Suh
SS
, Ladetto
M
, Kuehl
M
, Palumbo
T
, Drandi
D
, Taccioli
C
, Zanesi
N
, Alder
H
, Hagan
JP
, Munker
R
, Volinia
S
and Boccadoro
M.
MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis.
Proc Natl Acad Sci U S A.
2008;
105:
12885
-12890.
[PubMed]
.
-
23.
Sutherland
KD
, Lindeman
GJ
, Choong
DY
, Wittlin
S
, Brentzell
L
, Phillips
W
, Campbell
IG
and Visvader
JE.
Differential hyper-methylation of SOCS genes in ovarian and breast carcinomas.
Oncogene.
2004;
23:
7726
-7733.
[PubMed]
.
-
24.
Weniger
MA
, Melzner
I
, Menz
CK
, Wegener
S
, Bucur
AJ
, Dorsch
K
, Mattfeldt
T
, Barth
TF
and Moller
P.
Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation.
Oncogene.
2006;
25:
2679
-2684.
[PubMed]
.
-
25.
Yoshikawa
H
, Matsubara
K
, Qian
GS
, Jackson
P
, Groopman
JD
, Manning
JE
, Harris
CC
and Herman
JG.
SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity.
Nat Genet.
2001;
28:
29
-35.
[PubMed]
.
-
26.
Evan
GI
and d'Adda
di Fagagna F.
Cellular senescence: hot or what.
Curr Opin Genet Dev.
2009;
19:
25
-31.
[PubMed]
.
-
27.
Yu
H
and Jove
R.
The STATs of cancer--new molecular targets come of age.
Nat Rev Cancer.
2004;
4:
97
-105.
[PubMed]
.
-
28.
Mallette
FA
, Gaumont-Leclerc
MF
and Ferbeyre
G.
The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence.
Genes Dev.
2007;
21:
43
-48.
[PubMed]
.
-
29.
Mallette
FA
, Gaumont-Leclerc
MF
, Huot
G
and Ferbeyre
G.
Myc down-regulation as a mechanism to activate the Rb pathway in STAT5A-induced senescence.
J Biol Chem.
2007;
282:
34938
-34944.
[PubMed]
.
-
30.
Mallette
FA
, Moiseeva
O
, Calabrese
V
, Mao
B
, Gaumont-Leclerc
MF
and Ferbeyre
G.
Transcriptome analysis and tumor suppressor requirements of STAT5-induced senescence.
Ann N Y Acad Sci.
2010;
1197:
142
-151.
[PubMed]
.
-
31.
de Stanchina
E
, McCurrach
ME
, Zindy
F
, Shieh
SY
, Ferbeyre
G
, Samuelson
AV
, Prives
C
, Roussel
MF
, Sherr
CJ
and Lowe
SW.
E1A signaling to p53 involves the p19(ARF) tumor suppressor.
Genes Dev.
1998;
12:
2434
-2442.
[PubMed]
.
-
32.
Zindy
F
, Eischen
CM
, Randle
DH
, Kamijo
T
, Cleveland
JL
, Sherr
CJ
and Roussel
MF.
Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization.
Genes Dev.
1998;
12:
2424
-2433.
[PubMed]
.
-
33.
Wei
W
, Hemmer
RM
and Sedivy
JM.
Role of p14(ARF) in replicative and induced senescence of human fibroblasts.
Mol Cell Biol.
2001;
21:
6748
-6757.
[PubMed]
.
-
34.
Calabrese
V
, Mallette
FA
, Deschenes-Simard
X
, Ramanathan
S
, Gagnon
J
, Moores
A
, Ilangumaran
S
and Ferbeyre
G.
SOCS1 links cytokine signaling to p53 and senescence.
Mol Cell.
2009;
36:
754
-767.
[PubMed]
.
-
35.
de Stanchina
E
, Querido
E
, Narita
M
, Davuluri
RV
, Pandolfi
PP
, Ferbeyre
G
and Lowe
SW.
PML is a direct p53 target that modulates p53 effector functions.
Mol Cell.
2004;
13:
523
-535.
[PubMed]
.
-
36.
Bourdeau
V
, Baudry
D
and Ferbeyre
G.
PML links aberrant cytokine signaling and oncogenic stress to cellular senescence.
Front Biosci.
2009;
14:
475
-485.
[PubMed]
.
-
37.
Ferbeyre
G
, de Stanchina
E
, Querido
E
, Baptiste
N
, Prives
C
and Lowe
SW.
PML is induced by oncogenic ras and promotes premature senescence.
Genes Dev.
2000;
14:
2015
-2027.
[PubMed]
.
-
38.
Pearson
M
, Carbone
R
, Sebastiani
C
, Cioce
M
, Fagioli
M
, Saito
S
, Higashimoto
Y
, Appella
E
, Minucci
S
, Pandolfi
PP
and Pelicci
PG.
PML regulates p53 acetylation and premature senescence induced by oncogenic Ras.
Nature.
2000;
406:
207
-210.
[PubMed]
.
-
39.
Cleghon
V
and Morrison
DK.
Raf-1 interacts with Fyn and Src in a non-phosphotyrosine-dependent manner.
J Biol Chem.
1994;
269:
17749
-17755.
[PubMed]
.
-
40.
Park
I
, Chung
J
, Walsh
CT
, Yun
Y
, Strominger
JL
and Shin
J.
Phosphotyrosine-independent binding of a 62-kDa protein to the src homology 2 (SH2) domain of p56lck and its regulation by phosphorylation of Ser-59 in the lck unique N-terminal region.
Proc Natl Acad Sci U S A.
1995;
92:
12338
-12342.
[PubMed]
.
-
41.
Roe
JS
, Kim
H
, Lee
SM
, Kim
ST
, Cho
EJ
and Youn
HD.
p53 stabilization and transactivation by a von Hippel-Lindau protein.
Mol Cell.
2006;
22:
395
-405.
[PubMed]
.
-
42.
Bischof
O
, Schwamborn
K
, Martin
N
, Werner
A
, Sustmann
C
, Grosschedl
R
and Dejean
A.
The E3 SUMO ligase PIASy is a regulator of cellular senescence and apoptosis.
Mol Cell.
2006;
22:
783
-794.
[PubMed]
.
-
43.
Kahyo
T
, Nishida
T
and Yasuda
H Involvement of PIAS1 in the sumoylation of tumor suppressor p53.
Mol Cell.
2001;
8:
713
-718.
[PubMed]
.
-
44.
Megidish
T
, Xu
JH
and Xu
CW.
Activation of p53 by protein inhibitor of activated Stat1 (PIAS1).
J Biol Chem.
2002;
277:
8255
-8259.
[PubMed]
.
-
45.
Bartkova
J
, Rezaei
N
, Liontos
M
, Karakaidos
P
, Kletsas
D
, Issaeva
N
, Vassiliou
LV
, Kolettas
E
, Niforou
K
, Zoumpourlis
VC
, Takaoka
M
, Nakagawa
H
and Tort
F.
Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints.
Nature.
2006;
444:
633
-637.
[PubMed]
.
-
46.
Di Micco
R
, Fumagalli
M
, Cicalese
A
, Piccinin
S
, Gasparini
P
, Luise
C
, Schurra
C
, Garre
M
, Nuciforo
PG
, Bensimon
A
, Maestro
R
, Pelicci
PG
and d'Adda
di Fagagna F.
Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication.
Nature.
2006;
444:
638
-642.
[PubMed]
.
-
47.
Mallette
FA
and Ferbeyre
G.
The DNA damage signaling pathway connects oncogenic stress to cellular senescence.
Cell Cycle.
2007;
6:
1831
-1836.
[PubMed]
.
-
48.
Galanty
Y
, Belotserkovskaya
R
, Coates
J
, Polo
S
, Miller
KM
and Jackson
SP.
Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks.
Nature.
2009;
462:
935
-939.
[PubMed]
.
-
49.
Morris
JR
, Boutell
C
, Keppler
M
, Densham
R
, Weekes
D
, Alamshah
A
, Butler
L
, Galanty
Y
, Pangon
L
, Kiuchi
T
, Ng
T
and Solomon
E.
The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress.
Nature.
2009;
462:
886
-890.
[PubMed]
.
-
50.
Coppe
JP
, Patil
CK
, Rodier
F
, Sun
Y
, Munoz
DP
, Goldstein
J
, Nelson
PS
, Desprez
PY
and Campisi
J.
Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor.
PLoS Biol.
2008;
6:
2853
-2868.
[PubMed]
.
-
51.
Kuilman
T
, Michaloglou
C
, Vredeveld
LC
, Douma
S
, van
Doorn R
, Desmet
CJ
, Aarden
LA
, Mooi
WJ
and Peeper
DS.
Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network.
Cell.
2008;
133:
1019
-1031.
[PubMed]
.
-
52.
Rodier
F
, Coppe
JP
, Patil
CK
, Hoeijmakers
WA
, Munoz
DP
, Raza
SR
, Freund
A
, Campeau
E
, Davalos
AR
and Campisi
J.
Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion.
Nat Cell Biol.
2009;
11:
973
-979.
[PubMed]
.
-
53.
Acosta
JC
, O'Loghlen
A
, Banito
A
, Guijarro
MV
, Augert
A
, Raguz
S
, Fumagalli
M
, Da
Costa M
, Brown
C
, Popov
N
, Takatsu
Y
, Melamed
J
and d'Adda
di Fagagna F.
Chemokine signaling via the CXCR2 receptor reinforces senescence.
Cell.
2008;
133:
1006
-1018.
[PubMed]
.
-
54.
Wajapeyee
N
, Serra
RW
, Zhu
X
, Mahalingam
M
and Green
MR.
Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7.
Cell.
2008;
132:
363
-374.
[PubMed]
.
-
55.
Kortlever
RM
, Higgins
PJ
and Bernards
R.
Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence.
Nat Cell Biol.
2006;
8:
877
-884.
[PubMed]
.
-
56.
Xue
W
, Zender
L
, Miething
C
, Dickins
RA
, Hernando
E
, Krizhanovsky
V
, Cordon-Cardo
C
and Lowe
SW.
Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas.
Nature.
2007;
445:
656
-660.
[PubMed]
.
-
57.
Bekker-Jensen
S
, Rendtlew
Danielsen J
, Fugger
K
, Gromova
I
, Nerstedt
A
, Lukas
C
, Bartek
J
, Lukas
J
and Mailand
N.
HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes.
Nat Cell Biol.
2010;
12:
80
-86; sup pp 81-12.
[PubMed]
.
-
58.
Doil
C
, Mailand
N
, Bekker-Jensen
S
, Menard
P
, Larsen
DH
, Pepperkok
R
, Ellenberg
J
, Panier
S
, Durocher
D
, Bartek
J
, Lukas
J
and Lukas
C.
RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins.
Cell.
2009;
136:
435
-446.
[PubMed]
.
-
59.
Huen
MS
, Grant
R
, Manke
I
, Minn
K
, Yu
X
, Yaffe
MB
and Chen
J.
RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly.
Cell.
2007;
131:
901
-914.
[PubMed]
.
-
60.
Kolas
NK
, Chapman
JR
, Nakada
S
, Ylanko
J
, Chahwan
R
, Sweeney
FD
, Panier
S
, Mendez
M
, Wildenhain
J
, Thomson
TM
, Pelletier
L
, Jackson
SP
and Durocher
D Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase.
Science.
2007;
318:
1637
-1640.
[PubMed]
.
-
61.
Mailand
N
, Bekker-Jensen
S
, Faustrup
H
, Melander
F
, Bartek
J
, Lukas
C
and Lukas
J.
RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins.
Cell.
2007;
131:
887
-900.
[PubMed]
.
-
62.
Stewart
GS
, Panier
S
, Townsend
K
, Al-Hakim
AK
, Kolas
NK
, Miller
ES
, Nakada
S
, Ylanko
J
, Olivarius
S
, Mendez
M
, Oldreive
C
, Wildenhain
J
and Tagliaferro
A.
The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage.
Cell.
2009;
136:
420
-434.
[PubMed]
.
-
63.
Koelsche
C
, Strebovsky
J
, Baetz
A
and Dalpke
AH.
Structural and functional analysis of a nuclear localization signal in SOCS1.
Mol Immunol.
2009;
46:
2474
-2480.
[PubMed]
.
-
64.
Baetz
A
, Koelsche
C
, Strebovsky
J
, Heeg
K
and Dalpke
AH.
Identification of a nuclear localization signal in suppressor of cytokine signaling 1.
FASEB J.
2008;
22:
4296
-4305.
[PubMed]
.
-
65.
Boyd
SD
, Tsai
KY
and Jacks
T.
An intact HDM2 RING-finger domain is required for nuclear exclusion of p53.
Nat Cell Biol.
2000;
2:
563
-568.
[PubMed]
.
-
66.
Geyer
RK
, Yu
ZK
and Maki
CG.
The MDM2 RING-finger domain is required to promote p53 nuclear export.
Nat Cell Biol.
2000;
2:
569
-573.
[PubMed]
.
-
67.
Li
M
, Brooks
CL
, Wu-Baer
F
, Chen
D
, Baer
R
and Gu
W.
Mono- versus polyubiquitination: differential control of p53 fate by Mdm2.
Science.
2003;
302:
1972
-1975.
[PubMed]
.
-
68.
Sekimoto
T
, Imamoto
N
, Nakajima
K
, Hirano
T
and Yoneda
Y.
Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1.
EMBO J.
1997;
16:
7067
-7077.
[PubMed]
.
-
69.
Limnander
A
, Danial
NN
and Rothman
PB.
v-Abl signaling disrupts SOCS-1 function in transformed pre-B cells.
Mol Cell.
2004;
15:
329
-341.
[PubMed]
.
-
70.
Chen
JL
, Limnander
A
and Rothman
PB.
Pim-1 and Pim-2 kinases are required for efficient pre-B-cell transformation by v-Abl oncogene.
Blood.
2008;
111:
1677
-1685.
[PubMed]
.
-
71.
Chen
XP
, Losman
JA
, Cowan
S
, Donahue
E
, Fay
S
, Vuong
BQ
, Nawijn
MC
, Capece
D
, Cohan
VL
and Rothman
P.
Pim serine/threonine kinases regulate the stability of Socs-1 protein.
Proc Natl Acad Sci U S A.
2002;
99:
2175
-2180.
[PubMed]
.
-
72.
Srsen
V
, Gnadt
N
, Dammermann
A
and Merdes
A.
Inhibition of centrosome protein assembly leads to p53-dependent exit from the cell cycle.
J Cell Biol.
2006;
174:
625
-630.
[PubMed]
.
-
73.
Trinei
M
, Giorgio
M
, Cicalese
A
, Barozzi
S
, Ventura
A
, Migliaccio
E
, Milia
E
, Padura
IM
, Raker
VA
, Maccarana
M
, Petronilli
V
, Minucci
S
and Bernardi
P.
A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis.
Oncogene.
2002;
21:
3872
-3878.
[PubMed]
.
-
74.
White
DE
, Negorev
D
, Peng
H
, Ivanov
AV
, Maul
GG
and Rauscher
FJ.
3rd KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions.
Cancer Res.
2006;
66:
11594
-11599.
[PubMed]
.
-
75.
Yamauchi
T
, Negoro
E
, Kishi
S
, Takagi
K
, Yoshida
A
, Urasaki
Y
, Iwasaki
H
and Ueda
T.
Intracellular cytarabine triphosphate production correlates to deoxycytidine kinase/cytosolic 5'-nucleotidase II expression ratio in primary acute myeloid leukemia cells.
Biochem Pharmacol.
2009;
77:
1780
-1786.
[PubMed]
.
-
76.
Letsas
KP
, Frangou-Lazaridis
M
, Skyrlas
A
, Tsatsoulis
A
and Malamou-Mitsi
V.
Transcription factor-mediated proliferation and apoptosis in benign and malignant thyroid lesions.
Pathol Int.
2005;
55:
694
-702.
[PubMed]
.
-
77.
Wang
JY
, Sarker
AH
, Cooper
PK
and Volkert
MR.
The single-strand DNA binding activity of human PC4 prevents mutagenesis and killing by oxidative DNA damage.
Mol Cell Biol.
2004;
24:
6084
-6093.
[PubMed]
.
-
78.
Mortusewicz
O
, Roth
W
, Li
N
, Cardoso
MC
, Meisterernst
M
and Leonhardt
H.
Recruitment of RNA polymerase II cofactor PC4 to DNA damage sites.
J Cell Biol.
2008;
183:
769
-776.
[PubMed]
.