



Introduction
In a recent issue of Aging, Dubrovsky et al. [1] describe the effects of disruption of the Clock gene on lifespan and health in mice. They report that CLOCK-deficient mice have reduced average and maximum lifespan, and have increased incidence of dermatitis and cataracts as the animals age [1]. Notably, however, targeted disruption of the Clock gene does not lead to the same constellation of phenotypes as seen in mice with disruption of other genes critical for circadian clock function (Tables 1 and 2). In this Review, we summarize and discuss potential reasons for the gene-specific effects of circadian clock gene disruption. Why do these mouse models differ? And, what do these differences tell us about the role of the circadian clock in the regulation of physiology, healthy aging, and responses to genotoxic stress? Before addressing these questions, we will describe the circadian clock mechanism and the circadian clock gene hypothesis of cancer/aging.
Table 1. Rhythm and aging phenotypes of mice with mutations in genes contributing to the transcriptional activator complex
| ClockΔ19/Δ19 | Clock−/− | Npas2m/m | Clock−/−;Npas2m/m | Bmal1−/− | |
|---|---|---|---|---|---|
| Circadian Behavior: | |||||
| Rhythmic in DD? | Varies [123,141-144] | Yes [4, 117] | Yes [4, 145] | No [4] | No [106] |
| Period length | Long [~28 hr] to AR | Slightly shorter | Slightly shorter | N/A - Arrhythmic | N/A - Arrhythmic |
| Ex vivo Rhythms: | |||||
| SCN slice | Arrhythmic [124] | Rhythmic [4, 118] | Rhythmic [118] | Arrhythmic [118] | Arrhythmic [115, 124; but see 147] |
| Peripheral tissues | Arrhythmic [115, 124] | Arrhythmic [118] | Rhythmic [118] | Arrhythmic [118] | Arrhythmic [115, 147] |
| Aging Phenotypes | |||||
| Lifespan | Reduced (Female) Normal (Male) [49] | Reduced (Both) [1] | Seems OK (Both) [our unpublished] | Reduced (Both) [116] | Reduced (Both) [50, 107, 111] |
| Body weight | Increased [148] | Normal [1] | N.D. | Reduced [our unpublished] | Reduced [50] |
| Organ weights | Normal [148] | Normal [1] | N.D. | N.D. | Most Reduced [50] |
| Cataract incidence | N.D. | Increased [1] | N.D. | N.D. | Increased [50] |
| Dermatitis | N.D. | Increased [1] | N.D. | N.D. | Normal [48] |
| Ectopic calcification | No [our unpublished] | No [116] | No [116] | Yes [116] | Yes [108, 112] |
| Reproduction | Modest decrease [121, 122, 150-153] | Modest decrease [our unpublished] | Seems OK [4, 145, our unpublished] | Sterile [our unpublished] | Sterile [109, 110] |
| Tumor incidence | Normal [49] | N.D. | N.D. | N.D. | N.D. |
| Response to Irradiation | |||||
| Lifespan | Reduced (Female) Normal (Male) [49] | N.D. | N.D. | N.D. | N.D. |
| Body weight | Reduced (Both) [49] | N.D. | N.D. | N.D. | N.D. |
| Organ weights | Some Reduced [F]; Normal (Male) [49] | N.D. | N.D. | N.D. | N.D. |
| Cataract incidence | Increased (Both) [49] | N.D. | N.D. | N.D. | N.D. |
| Dermatitis | N.D. | N.D. | N.D. | N.D. | N.D. |
| Tumor incidence | Normal [49] | N.D. | N.D. | N.D. | N.D. |
Table 2. Rhythm and aging phenotypes of mice with mutations in genes contributing to the circadian repressor complex
| Cry1−/− | Cry2−/− | Cry1−/−; Cry2−/− | Per1−/− | Per2m/m | Per1−/−; Per2m/m | |
|---|---|---|---|---|---|---|
| Circadian behavior: | ||||||
| Rhythmic in DD? | Yes [7, | Yes [7, 8] | No [7, 8] | Varies [6, 9, 154] | Varies [6, 9, 156] | No [6, 9] |
| Period length | Shorter | Longer | N/A - Arrhythmic | Short or Normal | Short to Normal | N/A - Arrhythmic |
| Ex vivo Rhythms: | ||||||
| SCN slice | Rhythmic [146] | Rhythmic [146] | Arrhythmic [146] | Rhythmic [146] Arrhythmic [155] | Rhythmic [158] | N.D. |
| Peripheral tissues | Arrhythmic [146] | Rhythmic [146] | Arrhythmic [146] | Arrhythmic [146] | Varies [158] | N.D. |
| Aging parameters: | ||||||
| Lifespan | N.D. | N.D. | N.D. | N.D. | Decreased | N.D. |
| Body weight | N.D. | Normal [162] | Decreased [159] Normal [163] | Decreased [160, 161] | Increased [160, 161] | Normal [163] |
| Organ weights | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Cataract incidence | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Dermatitis | Increased [48] | N.D. | Increased [48] | N.D. | Normal [48] | Normal [48] |
| Ectopic calcification | No | No | No | No | No | No |
| Bone mass | N.D. | Increased [162] | Increased [163] | Normal [163] | Normal [163] | Increased [162, 163] |
| Reproduction | Seems OK (Both) [our unpublished] | Seems OK (Both) [our unpublished] | Seems OK (Both) [our unpublished] | Seems OK (Male) [our unpublished] Reduced (Female) [161] | Seems OK (Male) [our unpublished] Reduced (Female) [161] | Seems OK (Male) [our unpublished] Reduced (Female) [our unpublished] |
| Tumor incidence | Increased [48] | N.D. | Normal [23] Increased [48] | N.D. | Increased [46, 48] | Increased [48] |
| Response to Irradiation | ||||||
| Lifespan | N.D. | N.D. | Normal [23] Reduced [48] | N.D. | Reduced [46, 48] | Reduced [48] |
| Body weight | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Organ weights | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Cataract incidence | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Dermatitis | Small Increase [48] | N.D. | Increased [48] | N.D. | Normal [48] | Increased [48] |
| Tumor incidence | Increased [48] | N.D. | Normal [23] Increased [48] | N.D. | Increased [46,48] | Increased [48] |
| Cancer-prone model | p53−/− | APCmin/+ | ||||
| Cancer incidence | N.D. | N.D. | Decreased [51] | N.D. | Increased [47] | N.D. |
| Lifespan | N.D. | N.D. | Increased [51] | N.D. | Decreased [47] | N.D. |
The circadian clock is based on a transcriptional-translational feedback loop
Many cell types contain cell-autonomous circadian clocks that measure 24 hours. These clocks impart rhythmicity on function in virtually every organ. The biological timekeeping mechanism is based on a negative feedback loop involving the rhythmic production, followed by protracted degradation, of protein complexes that shut off their own production. The alternation between transcriptional activation and transcriptional inhibition occurs with a cycle length of approximately 24 hours [for review see 2,3]. In mammals, at the core of this mechanism are genes called Clock and Bmal1 (also called Mop3 or Arntl) (Figure 1). The protein products of these genes, CLOCK and BMAL1, are basic helix-loop-helix-PAS (bHLH-PAS) domain-containing transcription factors. They are dimerization partners of central importance to the function of the circadian clock, although NPAS2, another bHLH-PAS protein, can substitute for CLOCK as partner for BMAL1 in neurons of the suprachiasmatic nucleus (SCN) that control locomotor activity rhythms [4,5]. Proteins critical for closing the negative feedback loop include the products of the Per1 and Per2 genes, and the Cry1 and Cry2 genes; within these gene families there is redundancy of function, such that disruption of both Per1 and Per2, or both Cry1 and Cry2, is necessary to completely disrupt circadian rhythmicity [6-9]. Because these mutant lines provide multiple ways to disrupt circadian clock function, one might expect that a coherent picture of the role of the circadian clock in aging and the response to genotoxic stress would result. However, these mutant lines differ in their aging-related phenotypes, as well as having subtle differences in their circadian clock function.
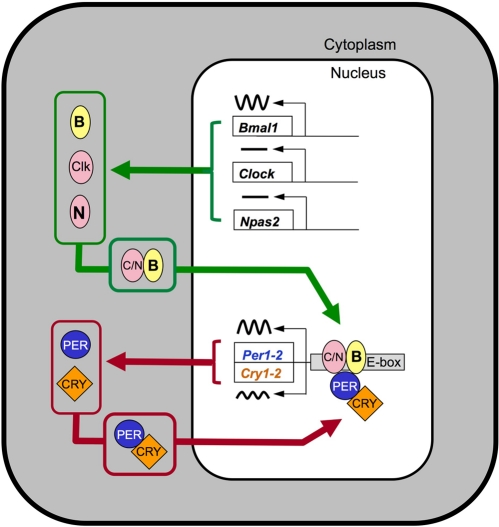
Figure 1. Model illustrating the core transcriptional-translational feedback loop underlying circadian rhythmicity. The components of the transcriptional activator complex, CLOCK, BMAL1, and (within SCN) NPAS2 form heterodimers that activate transcription of E-box containing genes, including the negative regulatory PER and CRY proteins. The PER-CRY protein complexes accumulate in the nucleus and block the activity of the activator complex. The alternation between the activation phase and the repression phase has a cycle length of near 24 hours. For simplicity, key events including posttranslational modifications and the generation of cascades of transcription factors leading to rhythmicity of tissue-specific “output” genes are not shown. For a more detailed model, see [2].
The circadian clock gene hypotheses of cancer and aging, corollaries and caveats
A great deal of experimental effort has addressed the role of the circadian clock genes in aging and especially in cancer-related phenotypes. The hypothesis being tested is usually that a circadian clock gene functions as a tumor suppressor gene, playing a role in retarding cancer development (or aging). More specifically, the hypothesis is that “circadian genes are involved in (cancer-related) biological pathways such as cell proliferation and apoptosis by controlling the expression of tumor suppressor genes, cell cycle genes, and genes encoding caspases and transcription factors” [10]. The basis for this hypothesis is that there are interactions between the cell cycle and circadian cycle, and dysregulation of the circadian clock is then thought to lead to dysregulation of cell cycle control [10].
Corollaries (with emphasis on cancer) are:
mutations of circadian clock genes will be associated with altered rates of cancer; circadian “clock gene” and protein expression levels will be altered in tumors; mutant mice with disrupted circadian clock genes will have altered (increased) cancer incidence under basal aging conditions, following gamma-irradiation, and in cancer-prone models.
Studies in support of this hypothesis are plentiful, as reviewed in greater detail below. Several studies indicate that circadian clock genes and their products contribute to regulation of cell cycle genes, apoptosis, and gene expression, and indicate molecular mechanisms by which these processes might be affected by clock gene products [11-21] [for recent reviews, see 22-33]. Many studies report altered (usually decreased) levels of clock gene expression in tumors [34-39; but see 40,41]. Conversely, in experimental models, overexpression of clock genes in tumors or in cancer cell lines reduces tumor growth [11, 42-45]. Mice homozygous for disruption of circadian clock genes that lead to loss of rhythmicity have many phenotypic abnormalities. Among these phenotypes are increases in basal cancer incidence or in the incidence of cancer following genotoxic stress or in genetically cancer-prone models [46-50; but see 51]. Similarly, knocking down the expression level of circadian clock genes can promote cancer growth [19, 52-55; but see 56, 57]. In humans, genome-wide association studies and more targeted studies have identified allelic variants, mutations, and epigenetic modifications that are associated with differences in risk of cancer [58-70]. Collectively, then, do these studies provide strong evidence that the circadian clock, and circadian rhythmicity, regulate aging and cancer? Remarkably, no. While the contribution of several circadian clock genes to cancer defense seem well-established, the contribution of the circadian clock is much less clear.
Functional impact of mutant alleles
In epidemiological studies, the circadian clock gene hypothesis of cancer proposes that genetic variations in clock genes are likely to be associated with individual susceptibility to cancer [62]. A more specific formulation of the hypothesis is that “circadian disruption may be a novel risk factor and that genetic determinants for circadian rhythms may play a role in… tumorigenesis” [59], which emphasizes the interaction of genetic predisposition based in the circadian clock genes with environmental influences (especially shift work and light exposure at night, which can disrupt diurnal rhythms; see below). Several epidemiological studies have been conducted, examining allele frequencies for polymorphisms in and around clock genes in association with different types of cancer. Many positive associations have been reported, both in targeted analyses and in genome-wide association studies [58-70].
An important factor for interpreting epidemiological studies is the observation in mutant mice that null alleles of circadian clock genes are usually recessive; that is, animals heterozygous for a loss-of-function allele have little or no deficit in overt circadian rhythms. Furthermore, within several of the circadian gene families, there is functional redundancy between closely related genes [Reviewed in [2]]. Mutations that lead to loss of circadian clock function have global effects on systemic physiology and metabolism, endocrine function and behavior. The functional impact of single nucleotide polymorphisms (SNP's) within or near circadian clock genes on the maintenance of rhythmicity is much less clear. It is likely that most SNPs near circadian genes do not lead to demonstrable changes in circadian oscillator function, either centrally or peripherally. Circadian clock genes may be influencing disease susceptibility due to their pleiotropic activities on gene expression or involvement in other pathways, rather than through their involvement in circadian clock function. As with GWAS studies in most fields, progression from identification of loci to elucidation of cellular and biochemical mechanisms is quite difficult.
Gene expression rhythmicity or gene expression level?
Microarray studies show that from 5-10% of transcripts are expressed with 24-hour rhythmicity. The specific genes that are rhythmic vary from tissue to tissue [17, 71-82], but key, rate-limiting steps are often rhythmic [71,72]. Disrupting critical circadian clock genes not only disrupts rhythmicity of target gene expression, but it can also have dramatic consequences on the level of expression of many target genes [73. 76-77]. Furthermore, the manner in which the circadian feedback loop is disrupted often has differing effects on the resulting level of gene expression. So, one might expect that disrupting genes on the activational side of the transcriptional feedback loop (Clock/Bmal1/Npas2) might have effects on target gene expression that are opposite to the effects seen when disrupting the genes whose products form the repressor complex (Pers and Crys; in this case the activator complex may act unopposed (Figure 1)). (To use a household analogy, in the former case the furnace is broken so no heat is produced; in the latter case the thermostat is broken so the signal to turn off heat production is not given, and the room overheats. In both cases the system is broken, but the effects on steady state room temperature are opposite). An interesting distinction to keep in mind when considering phenotypes resulting from circadian gene disruption is whether the phenotype could be due to loss of rhythmicity of gene expression, versus due to the potentially disruption-specific effects on gene expression level. In the former case, we might expect that all mutations of circadian genes leading to loss of rhythmicity would have a similar phenotype, while in the latter case we can envision how disruption of different circadian genes could have opposite effects.
Environmental lighting, rhythms and disease
It is also worth pointing out that almost all studies of aging-related endpoints in circadian mutant mice have been conducted with animals housed in a light-dark cycle. Rhythmic lighting cycles can drive behavioral and physiological rhythmicity (to varying extents, depending on the genotype and circumstances) even in the absence of a functioning circadian clock (e.g., see Figure 2C). Thus, many studies are assessing the phenotype in the presence of LD- imposed rhythmicity. The phenotype of circadian mutant mice may be considerably more pronounced if they were to be studied after housing in constant dark conditions. (Constant light is rarely used as it can disrupt rhythms if too bright, and controlling light intensity across a group of cages is more difficult than maintaining consistency from cage to cage with darkness.) Two recent studies [48, 83] highlight the exacerbation of that can occur when circadian mutant mice are housed in constant darkness, compared to the same genotype housed in a standard light: dark cycle. Thus, circadian mutant mice are often studied under conditions that may not allow full expression of their genetic deficits. Nevertheless, as both a practical matter related to monitoring of health and with respect to relevance to human health and disease, studying mutant animals in a lighting cycle is appropriate.
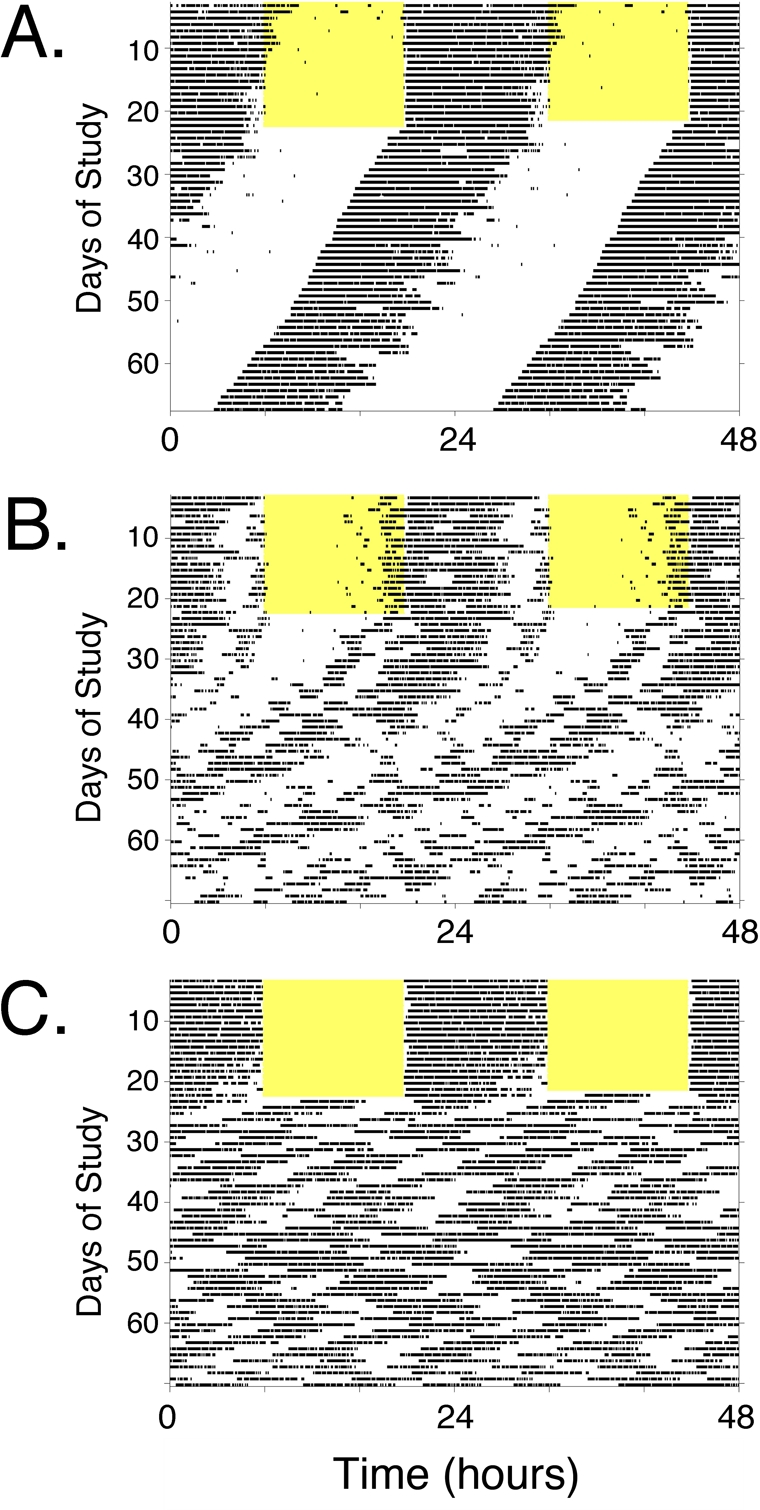
Figure 2. Locomotor activity rhythms of circadian mutant mice. Panels A-C, Double-plotted actograms illustrating types of locomotor activity patterns observed in wild-type and circadian mutant mice (see Tables 1 and 2). Each panel represents the locomotor activity pattern of a single mouse housed in a cage with a running wheel. Periods of voluntary wheel-running appear as dark bars within the grids. Each line of the record shows 48 hours of data across, and successive days are plotted below the first. Each animal was exposed to a standard 12 hour light: 12 hour dark lighting cycle (light period indicated by the yellow shading) for ~ 2 weeks. Then, the light-dark cycle was disabled and the animals were recorded in constant darkness (DD), which allows expression of the animals' endogenous rhythmicity. Visual analysis of the “actogram” allows easy perception of the cycle length (period length, represented by the slope of the line connecting activity onsets) and the robustness of the rhythm when the animal is in DD. (A) The actogram in Panel A is typical of a wild-type mouse. In the lighting cycle, activity is confined to the dark phase of the cycle. Upon entry into DD, the animal maintains robust rhythmicity with a cycle length of ~ 23.6 hours. This is seen as a leftward shift of activity onset each day by ~ 20 minutes. Lines of mice that would have similar activity records, with or without slight changes in circadian null mutations, CLOCK-deficient mice and NPAS2-deficient mice. Larger alterations in period length but with maintenance of robust rhythmicity is seen in CRY1-deficient mice, CRY2-deficient mice, and Clock Δ19/+ heterozygotes. (B) The actogram in Panel B is typical of a mouse with gradual loss of rhythmicity. The light-dark cycle synchronized activity to nighttime, and after transfer to DD, the animal expresses a “free-running” rhythm that gradually decreases in amplitude until arrhythmicity is reached. Mice homozygous for mutation of Per1 or Per2 have this phenotype (although in some studies these lines maintain rhythmicity and resemble Panel A). ClockΔ19/Δ19 mutant mice initially have long-period (~ 28 hr) rhythmicity and become arrhythmic within ~ 10 days in constant darkness. (C) The actogram in Panel C is typical of an animal with complete loss of circadian function. Note that the mouse appears to have rhythmic behavior when exposed to a light-dark cycle, due to suppression of wheel-running activity by ambient light. Following discontinuation of the light-dark cycle, however, the endogenous pattern (or lack there-of) becomes apparent. In this case, the animal immediately loses circa-24-hour rhythmicity. Despite loss of circadian rhythmicity, the animal still has periods of activity and rest, just now these intervals do not have the temporal organization on a 24-hour timescale. Short-period rhythms (4-6 hr cycles) predominate. This pattern is typical of line of mice referred to as arrhythmic, including Clock/Npas2-double-knockout (Clock−/−; Npas2m/m) mice, Bmal1−/− knockout, Per1/Per2 double knockout, and Cry1/Cry2 double-knockout mice.
Humans, generally, do not reside in constant darkness. Remarkably, however, several studies on cancer incidence among blind individuals reveal a reduced risk [84-90]. Cancer risk is lower when blindness is defined in the strictest sense (those with no capacity for light photoreception), even relative to those that are blind with light perception [80]. Such studies were motivated by the observation that exposure to light at night during shift work leads to circadian disruption and also significantly increases the risk of cancer [86, 87, 89]. Indeed, disrupted circadian rhythmicity caused by light exposure at night has been proposed to underly the increasing prevalence of breast and prostate cancers in industrialized nations. The International Agency for Research on Cancer (IARC) recently concluded that “shift-work that involves circadian disruption is probably carcinogenic to humans” based on “limited evidence in humans” and “sufficient evidence in experimental animals for the carcinogenicity of light during the daily dark period” [91]. Whether these effects are related to nocturnal suppression of melatonin production, as advocated by some, remains to be established; melatonin has anticancer activity in some circumstances [92]. It has been argued that light exposure at night is a predictor of adverse health outcomes, rather than a cause [93].
Several recent studies suggest that the process of repeatedly resetting one's clock is a stressor with adverse effects on health. Repeated exposure to shifts of the light-dark cycle (chronic jet lag) reduces lifespan in aged mice [94], alters the immune response to lipopolysacchiride challenge in young mice [95], accelerates liver carcinogenesis [96-98], and increases susceptibility to gastrointestinal damage by dextran sulfate [99]. Notably, some of these studies were conducted in strains of mice genetically incapable of producing melatonin [100], so shift-induced disruption of the (nonexistent) melatonin rhythm cannot be the mechanism underlying the adverse response. In other studies, lines of hamsters that differ in their free-running cycle length were found to differ in the development of cardiac and renal pathologies; animals housed in a light-dark cycle that did not match their endogenous cycle length had to undergo re-setting of their clock on a daily basis to maintain synchrony to the lighting cycle [101, 102]. Remarkably, in the hamster studies, each line did better when the lighting cycle matched its endogenous cycle length, and poorly (relative to the other line) when there is a mismatch. These animal studies suggest that environmental disruption of circadian rhythms has a significant adverse effect on health, including carcinogenesis and immune function. Circadian misalignment can also affect metabolic function in humans [103] with potential consequences on antioxidant defenses. Thus, there are important consequences of circadian rhythm disruption on aging and cancer.
Version control
The version of the circadian clock gene hypothesis being tested can vary greatly between studies, and authors are rarely explicit in articulating the version being tested, or its implications. Two extreme cases can make this point. Lee et al., [48] recently proposed that the central circadian clock in the suprachiasmatic nucleus of the hypothalamus coordinates the activity of peripheral clocks via the sympathetic nervous system, and in so doing optimizes anticancer mechanisms within peripheral cells. Their data suggest that any of a number of genetic lesions leading to loss of rhythmicity promote tumorigenesis, e.g., that the circadian clock reduces cancer risk through systemic mechanisms. Furthermore, as noted above, in this study maintaining animals in light-dark cycles masked the cancer phenotype [48]. At the other extreme are data that indicate that altering levels of clock gene expression affects proliferation in vitro; these effects cannot be related to systemic physiological rhythmicity, yet could still involve cellular rhythmicity (which persists in vitro; 104,105]. Data from both types of studies are relevant to the circadian-aging-cancer continuum, but these studies implicate different mechanisms and require different interpretations. Questions relevant to studies of circadian clock genes and cancer include:
- Is behavioral rhythmicity affected by the genetic lesion? - Is the circadian clock broken in an “on” or “off” state? - What impact does this gene have on expression of other genes and processes, and are these necessarily related to rhythmicity?
Prospectus
What does the future hold for studies of the role of circadian clock genes in aging, response to genotoxic stress, and cancer? Most obviously, filling in the gaps in Tables 1 and 2 is needed. How are the acute response to radiation and radiation-induced morbidity, cancer incidence, and mortality influenced by the absence of CLOCK? What are the causes of premature death in CLOCK-deficient mice? A striking preliminary conclusion from the studies discussed here is that lifespan and cancer incidence of circadian mutant lines under baseline conditions appear to be poorly predictive of each other or of responses to genotoxic stress and irradiation [22, 33, 49, 50].
Another line of investigation is to understand the molecular mechanisms underlying the age-related pathologies in CLOCK-deficient mice identified by Dubrovsky et al [1]. What does a higher incidence and earlier onset of dermatitis in CLOCK-deficient mice mean? Differences in aggression and interactions between cage-mates or in grooming could contribute [1], but in addition, the accelerated, age-dependent incidence of dermatitis in CLOCK-deficient mice could be due to loss of an important, local action of the CLOCK:BMAL1 dimer and/or circadian rhythms of gene expression. Fibroblasts, including human dermal fibroblasts, have tissue-autonomous molecular circadian rhythms [124, 125, 128-132]. Rhythmic clock gene expression in skin is disrupted in CRY-deficient or SCN-lesioned mice [133]. Hair follicle synchrony (on a ~ 3-week cycle length) is altered in CLOCKΔ19 and Bmal1−/− mice [134, 135], further indicating a role for circadian clock function in skin. Thus, disruption of circadian clock genes likely disrupts skin physiology by local actions. Alternatively, systemic hormonal, metabolic or nutritional factors may influence dermal integrity. Histological examination and assessment of constituent cell populations is necessary to relate the observation of increased dermatitis incidence in CLOCK-deficient mice to epithelial, mast, and dendritic cells, sebaceous glands and hair follicles.
Similarly, what does the higher incidence of cataracts in CLOCK-deficient mice mean? Cataracts are often taken as an indication of excessive reactive oxygen species (ROS) and oxidative stress, and indeed, in BMAL1-deficient mice, cataracts are delayed by treatment with an antioxidant [107]. Does the increased age-dependent incidence of cataracts in CLOCK-deficient mice reflect whole-body ROS status, or local events such as oxygen tension and ion transport in the lens epithelia? In future studies, it may be possible to distinguish systemic, metabolic influences from local effects by local manipulation of these genes, using conditional alleles of these genes (in which critical exons are flanked by loxP sites) and tissue-specific drivers for Cre recombinase. Tissue-specific gene disruption is beginning to be employed in studies of circadian rhythms [78, 82, 114, 115, 117].
When discussing circadian rhythms and oxidative stress, melatonin may come to mind. As noted above, melatonin is produced in a circadian rhythm, with levels elevated at night, and the hormone is widely investigated with respect to its antioxidant properties following administration at pharmacological levels [136, 137]. The role of endogenous melatonin in antioxidant defense is unclear. In the present context, however, it is clear that melatonin can play no role: C57Bl/6J mice do not produce pineal melatonin [100].
Another interesting area for study will to be to determine the extent to which NPAS2 substitutes for CLOCK, mitigating the impact of CLOCK deficiency (relative to the more profound effects of BMAL1-deficiency). In the SCN, NPAS2 maintains circadian rhythmicity in the absence of CLOCK [4]. In contrast, peripheral tissues are unable to maintain rhythmicity in the absence of CLOCK [111, 138, but see 139]. Mice lacking both CLOCK and NPAS2 have a profound phenotype, appearing very similar to mice lacking BMAL1 with respect to age-dependent weight loss, arthropathy, male and female sterility, and premature death [Table 1; our laboratory's unpublished data]. The very profound accelerated aging phenotype of these double-knockout mice, especially when compared to mice lacking CLOCK alone, suggests an important, redundant contribution of NPAS2—but where? In CLOCK-deficient mice, pathologies may be restricted to those tissues that have naturally low levels of NPAS2 expression, so in these tissues there is a breakdown of redundancy. Alternatively, the loss of systemic rhythmicity may interact in important ways. An exciting prospect is that the conditional (floxed) alleles of Clock (in an NPAS2-deficient background) or conditional alleles of Bmal1 will allow identification of sites and mechanisms of action of premature spontaneous aging and of accelerated aging induced by genotoxic stress. Furthermore, the availability of mouse lines in which expression of CLOCK or BMAL1 can be induced with anatomical and temporal control [112, 140] allow complementary genetic manipulations to gene disruption strategies.
Conclusions
While the studies conducted to date do not, in our opinion, generate a clear conclusion about the contribution of circadian rhythmicity to normal aging, several circadian clock genes are clearly important in regulating lifespan, cancer susceptibility and the cell cycle. Further studies involving manipulation of circadian clock genes, and manipulating the environmental housing conditions in which animals are studied, have much to offer in the study of aging.
Acknowledgments
Work in the authors' laboratory was supported by National Institutes of Health Award R01 NS056125 from the National Institutes of Neurological Diseases and Stroke (to DRW). DRW is a member of the University of Massachusetts Medical School Diabetes and Endocrinology Research Center (DK 32520). The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the sponsoring agencies.
Conflicts of Interest
The authors of this paper declare no conflict of interests.
References
- 1. Dubrovsky YV, Samsa WE, Kondratov RV. Deficiency of circadian protein CLOCK reduces lifespan and increases age–related cataract development in mice. Aging (Albany). 2010; 2: 936 -944. .
- 2. Weaver DR and Reppert SM. Circadian timekeeping Fundamental Neuroscience. Squire L.R., Berg D., Bloom F.E., du Lac S., Ghosh A., Spitzer N.C.. San Diego Academic Press 931 -958. 2008; .
- 3. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: Organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010; 72: 517 -549. [PubMed] .
- 4. DeBruyne JP, Weaver DR, Reppert SM. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat Neurosci. 2007; 10: 543 -545. [PubMed] .
- 5. DeBruyne JP. Oscillating perceptions: The ups and downs of the CLOCK protein in the mouse circadian system. J Genet. 2008; 87: 437 -446. [PubMed] .
- 6. Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, Weaver DR. Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron. 2001; 30: 525 -36. [PubMed] .
- 7. van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, de Wit J, Verkerk A, Eker AP, van Leenen D, Buijs R, Bootsma D, Hoeijmakers JH, et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999; 398: 627 -630. [PubMed] .
- 8. Vitaterna MH, Selby CP, Todo T, Niwa H, Thompson C, Fruechte EM, Hitomi K, Thresher RJ, Ishikawa T, Miyazaki J, Takahashi JS, Sancar A. Differential regulation of mammalian Period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc Natl Acad Sci U S A. 1999; 96: 12114 -12119. [PubMed] .
- 9. Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, Vaishnav S, Li Q, Sun ZS, Eichele G, Bradley A, Lee CC. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell. 2001; 105: 683 -694. [PubMed] .
- 10. Fu L and Lee CC. The circadian clock: Pacemaker and tumour suppressor. Nat Rev Cancer. 2003; 3: 350 -361. [PubMed] .
- 11. Cao Q, Gery S, Dashti A, Yin D, Zhou Y, Gu J, Koeffler HP. A role for the clock gene per1 in prostate cancer. Cancer Res. 2009; 69: 7619 -7625. [PubMed] .
- 12. Gery S, Virk RK, Chumakov K, Yu A, Koeffler HP. The clock gene Per2 links the circadian system to the estrogen receptor. Oncogene. 2007; 26: 7916 -7920. [PubMed] .
- 13. Jung-Hynes B and Ahmad N. SIRT1 controls circadian clock circuitry and promotes cell survival: A connection with age-related neoplasms. FASEB J. 2009; 23: 2803 -2809. [PubMed] .
- 14. Kang TH, Lindsey-Boltz LA, Reardon JT, Sancar A. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc Natl Acad Sci U S A. 2010; 107: 4890 -4895. [PubMed] .
- 15. Kang TH and Sancar A. Circadian regulation of DNA excision repair: Implications for chrono-chemotherapy. Cell Cycle. 2009; 8: 1665 -1667. [PubMed] .
- 16. Matsuo T., Yamaguchi S., Mitsui S., Emi A., Shimoda F., Okamura H.. 2003; Control mechanism of the circadian clock for timing of cell division in vivo. Science. 302: 255 -259. [PubMed] .
- 17. Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, Antoch MP, Walker JR, Esser KA, Hogenesch JB, Takahashi JS. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci U S A. 2007; 104: 3342 -3347. PMID: 17360649 [PubMed] .
- 18. Miyamoto N, Izumi H, Noguchi T, Nakajima Y, Ohmiya Y, Shiota M, Kidani A, Tawara A, Kohno K. Tip60 is regulated by circadian transcription factor clock and is involved in cisplatin resistance. J Biol Chem. 2008; 283: 18218 -18226. [PubMed] .
- 19. Mullenders J, Fabius AW, Madiredjo M, Bernards R, Beijersbergen RL. A large scale shRNA barcode screen identifies the circadian clock component ARNTL as putative regulator of the p53 tumor suppressor pathway. PLoS One. 2009; 4: e4798 [PubMed] .
- 20. Uchida Y, Hirayama J, Nishina H. A common origin: Signaling similarities in the regulation of the circadian clock and DNA damage responses. Biol Pharm Bull. 2010; 33: 535 -544. [PubMed] .
- 21. Gréchez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem. 2008; 283: 4535 -4542. [PubMed] .
- 22. Antoch MP, Kondratov RV, Takahashi JS. Circadian clock genes as modulators of sensitivity to genotoxic stress. Cell Cycle. 2005; 4: 901 -907. [PubMed] .
- 23. Gauger MA and Sancar A. Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res. 2005; 65: 6828 -6834. [PubMed] .
- 24. Sahar S and Sassone-Corsi P.. Circadian clock and breast cancer: A molecular link. Cell Cycle. 2007; 6: 1329 -1331. [PubMed] .
- 25. Borgs L, Beukelaers P, Vandenbosch R, Belachew S, Nguyen L, Malgrange B. Cell ‘circadian’ cycle: New role for mammalian core clock genes. Cell Cycle. 2009; 8: 832 -837. [PubMed] .
- 26. Hede K. Cancer and the circadian clock: Has the time finally come? J Natl Cancer Inst. 2009; 101: 550 -553. [PubMed] .
- 27. Wood PA, Yang X, Hrushesky WJ. Clock genes and cancer. Integr Cancer Ther. 2009; 8: 303 -308. [PubMed] .
- 28. Gery S and Koeffler HP. Circadian rhythms and cancer. Cell Cycle. 2010; 9: 1097 -1103. [PubMed] .
- 29. Khapre RV, Samsa WE, Kondratov RV. Circadian regulation of cell cycle: Molecular connections between aging and the circadian clock. Ann Med. 2010; 42: 404 -415. [PubMed] .
- 30. Morf J and Schibler U. Circadian cell-cycle progression: Cracking open the gate. Cell. 2010; 140: 458 -459. [PubMed] .
- 31. Rana S and Mahmood S. Circadian rhythm and its role in malignancy. J Circadian Rhythms. 2010; 8: 3 [PubMed] .
- 32. Sancar A, Lindsey-Boltz LA, Kang TH, Reardon JT, Lee JH, Ozturk N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010; 584: 2618 -2625. [PubMed] .
- 33. Antoch MP and Kondratov RV. Circadian proteins and genotoxic stress response. Circ Res. 2010; 106: 68 -78. [PubMed] .
- 34. Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ, Chang JG. Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis. 2005; 26: 1241 -1246. [PubMed] .
- 35. Kuo SJ, Chen ST, Yeh KT, Hou MF, Chang YS, Hsu NC, Chang JG. Disturbance of circadian gene expression in breast cancer. Virchows Arch. 2009; 454: 467 -474. [PubMed] .
- 36. Krugluger W, Brandstaetter A, Kallay E, Schueller J, Krexner E, Kriwanek S, Bonner E, Cross HS. Regulation of genes of the circadian clock in human colon cancer: Reduced period-1 and dihydropyrimidine dehydrogenase transcription correlates in high-grade tumors. Cancer Res. 2007; 67: 7917 -7922. [PubMed] .
- 37. Mostafaie N, Kallay E, Sauerzapf E, Bonner E, Kriwanek S, Cross HS, Huber KR, Krugluger W. Correlated downregulation of estrogen receptor beta and the circadian clock gene Per1 in human colorectal cancer. Mol Carcinog. 2009; 48: 642 -647. [PubMed] .
- 38. Winter SL, Bosnoyan-Collins L, Pinnaduwage D, Andrulis IL. Expression of the circadian clock genes Per1 and Per2 in sporadic and familial breast tumors. Neoplasia. 2007; 9: 797 -800. [PubMed] .
- 39. Xia HC, Niu ZF, Ma H, Cao SZ, Hao SC, Liu ZT, Wang F. Deregulated expression of the Per1 and Per2 in human gliomas. Can J Neurol Sci. 2010; 37: 365 -370. [PubMed] .
- 40. Geusz ME, Blakely KT, Hiler DJ, Jamasbi RJ. Elevated mPer1 gene expression in tumor stroma imaged through bioluminescence. Int J Cancer. 2010; 126: 620 -630. [PubMed] .
- 41. Sato F, Nagata C, Liu Y, Suzuki T, Kondo J, Morohashi S, Imaizumi T, Kato Y, Kijima H. PERIOD1 is an anti-apoptotic factor in human pancreatic and hepatic cancer cells. J Biochem. 2009; 146: 833 -838. [PubMed] .
- 42. Hua H, Wang Y, Wan C, Liu Y, Zhu B, Wang X, Wang Z, Ding JM. Inhibition of tumorigenesis by intratumoral delivery of the circadian gene mPer2 in C57BL/6 mice. Cancer Gene Ther. 2007; 14: 815 -818. [PubMed] .
- 43. Hua H, Wang Y, Wan C, Liu Y, Zhu B, Yang C, Wang X, Wang Z, Cornelissen-Guillaume G, Halberg F. Circadian gene mPer2 overexpression induces cancer cell apoptosis. Cancer Sci. 2006; 97: 589 -96. [PubMed] .
- 44. Miyazaki K, Wakabayashi M, Hara Y, Ishida N. Tumor growth suppression in vivo by overexpression of the circadian component, PER2. Genes Cells. 2010; 15: 351 -358. [PubMed] .
- 45. Oda A, Katayose Y, Yabuuchi S, Yamamoto K, Mizuma M, Shirasou S, Onogawa T, Ohtsuka H, Yoshida H, Hayashi H, Rikiyama T, Kim H, Choe Y, et al. Clock gene mouse period2 overexpression inhibits growth of human pancreatic cancer cells and has synergistic effect with cisplatin. Anticancer Res. 2009; 29: 1201 -1209. [PubMed] .
- 46. Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002; 111: 41 -50. [PubMed] .
- 47. Wood PA, Yang X, Taber A, Oh EY, Ansell C, Ayers SE, Al-Assaad Z, Carnevale K, Berger FG, Pena MM, Hrushesky WJ. Period 2 mutation accelerates ApcMin/+ tumorigenesis. Mol Cancer Res. 2008; 6: 1786 -1793. [PubMed] .
- 48. Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS One. 2010; 5: e10995 [PubMed] .
- 49. Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, Lee C, Nikitin AY. Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle. 2008; 7: 1197 -1204. [PubMed] .
- 50. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006; 20: 1868 -1873. [PubMed] .
- 51. Ozturk N, Lee JH, Gaddameedhi S, Sancar A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci U S A. 2009; 106: 2841 -2846. [PubMed] .
- 52. Yang X, Wood PA, Ansell CM, Quiton DF, Oh EY, Du-Quiton J, Hrushesky WJ. The circadian clock gene Per1 suppresses cancer cell proliferation and tumor growth at specific times of day. Chronobiol Int. 2009; 26: 1323 -1339. [PubMed] .
- 53. Yang X, Wood PA, Oh EY, Du-Quiton J, Ansell CM, Hrushesky WJ. Down regulation of circadian clock gene Period 2 accelerates breast cancer growth by altering its daily growth rhythm. Breast Cancer Res Treat. 2009; 117: 423 -31. [PubMed] .
- 54. Yang X, Wood PA, Ansell C, Hrushesky WJ. Circadian time-dependent tumor suppressor function of Period genes. Integr Cancer Ther. 2009; 8: 309 -316. [PubMed] .
- 55. Zeng ZL, Wu MW, Sun J, Sun YL, Cai YC, Huang YJ, Xian LJ. Effects of the biological clock gene Bmal1 on tumour growth and anti-cancer drug activity. J Biochem. 2010; 148: 319 -326. [PubMed] .
- 56. Sato F, Nagata C, Liu Y, Suzuki T, Kondo J, Morohashi S, Imaizumi T, Kato Y, Kijima H. PERIOD1 is an anti-apoptotic factor in human pancreatic and hepatic cancer cells. J Biochem. 2009; 146: 833 -838. [PubMed] .
- 57. Suzuki T, Sato F, Kondo J, Liu Y, Kusumi T, Fujimoto K, Kato Y, Sato T, Kijima H. Period is involved in the proliferation of human pancreatic MIA-PaCa2 cancer cells by TNF-alpha. Biomed Res. 2008; 29: 99 -103. [PubMed] .
- 58. Zhu Y, Brown HN, Zhang Y, Stevens RG, Zheng T. Period3 structural variation: a circadian biomarker associated with breast cancer in young women. Cancer Epidemiol Biomarkers Prev. 2005; 14: 268 -70. [PubMed] .
- 59. Zhu Y, Zheng T, Stevens RG, Zhang Y, Boyle P. Does ‘clock’ matter in prostate cancer? Cancer Epidemiol Biomarkers Prev. 2006; 15: 3 -5. [PubMed] .
- 60. Zhu Y, Leaderer D, Guss C, Brown HN, Zhang Y, Boyle P, Stevens RG, Hoffman A, Qin Q, Han X, Zheng T. Ala394Thr polymorphism in the clock gene NPAS2: A circadian modifier for the risk of non-hodgkin's lymphoma. Int J Cancer. 2007; 120: 432 -435. [PubMed] .
- 61. Zhu Y, Stevens RG, Leaderer D, Hoffman A, Holford T, Zhang Y, Brown HN, Zheng T. Non-synonymous polymorphisms in the circadian gene NPAS2 and breast cancer risk. Breast Cancer Res Treat. 2008; 107: 421 -425. [PubMed] .
- 62. Zhu Y, Stevens RG, Hoffman AE, Fitzgerald LM, Kwon EM, Ostrander EA, Davis S, Zheng T, Stanford JL. Testing the circadian gene hypothesis in prostate cancer: A population-based case-control study. Cancer Res. 2009; 69: 9315 -9322. [PubMed] .
- 63. Taniguchi H, Fernandez AF, Setien F, Ropero S, Ballestar E, Villanueva A, Yamamoto H, Imai K, Shinomura Y, Esteller M. Epigenetic inactivation of the circadian clock gene BMAL1 in hematologic malignancies. Cancer Res. 2009; 69: 8447 -8454. [PubMed] .
- 64. Alhopuro P, Bjorklund M, Sammalkorpi H, Turunen M, Tuupanen S, Bistrom M, Niittymaki I, Lehtonen HJ, Kivioja T, Launonen V, Saharinen J, Nousiainen K, Hautaniemi S, et al. Mutations in the circadian gene CLOCK in colorectal cancer. Mol Cancer Res. 2010; 8: 952 -960. [PubMed] .
- 65. Dai H, Zhang L, Cao M, Song F, Zheng H, Zhu X, Wei Q, Zhang W, Chen K. The role of polymorphisms in circadian pathway genes in breast tumorigenesis. Breast Cancer Res Treat. 2010; .
- 66. Hoffman AE, Zheng T, Stevens RG, Ba Y, Zhang Y, Leaderer D, Yi C, Holford TR, Zhu Y. Clock-cancer connection in non-Hodgkin's lymphoma: A genetic association study and pathway analysis of the circadian gene cryptochrome 2. Cancer Res. 2009; 69: 3605 -3613. [PubMed] .
- 67. Hoffman AE, Yi CH, Zheng T, Stevens RG, Leaderer D, Zhang Y, Holford TR, Hansen J, Paulson J, Zhu Y. CLOCK in breast tumorigenesis: Genetic, epigenetic, and transcriptional profiling analyses. Cancer Res. 2010; 70: 1459 -1468. [PubMed] .
- 68. Hoffman AE, Zheng T, Ba Y, Stevens RG, Yi CH, Leaderer D, Zhu Y. Phenotypic effects of the circadian gene Cryptochrome 2 on cancer-related pathways. BMC Cancer. 2010; 10: 110 [PubMed] .
- 69. Hoffman AE, Zheng T, Yi CH, Stevens RG, Ba Y, Zhang Y, Leaderer D, Holford T, Hansen J, Zhu Y. The core circadian gene Cryptochrome 2 influences breast cancer risk, possibly by mediating hormone signaling. Cancer Prev Res (Phila). 2010; 3: 539 -48. [PubMed] .
- 70. Yi C, Mu L, de la Longrais IA, Sochirca O, Arisio R, Yu H, Hoffman AE, Zhu Y, Katsaro D. The circadian gene NPAS2 is a novel prognostic biomarker for breast cancer. Breast Cancer Res Treat. 2010; 120: 663 -669. [PubMed] .
- 71. Akhtar RA, Reddy AB, Maywood ES, Clayton JD, King VM, Smith AG, Gant TW, Hastings MH, Kyriacou CP. Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol. 2002; 12: 540 -550. [PubMed] .
- 72. Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002; 109: 307 -320. [PubMed] .
- 73. Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002; 417: 78 -83. [PubMed] .
- 74. Kita Y, Shiozawa M, Jin W, Majewski RR, Besharse JC, Greene AS, Jacob HJ. Implications of circadian gene expression in kidney, liver and the effects of fasting on pharmacogenomic studies. Pharmacogenetics. 2002; 12: 55 -65. [PubMed] .
- 75. Duffield GE, Best JD, Meurers BH, Bittner A, Loros JJ, Dunlap JC. Circadian programs of transcriptional activation, signaling, and protein turnover revealed by microarray analysis of mammalian cells. Curr Biol. 2002; 12: 551 -557. [PubMed] .
- 76. Rudic RD, McNamara P, Reilly D, Grosser T, Curtis AM, Price TS, Panda S, Hogenesch JB, FitzGerald GA. Bioinformatic analysis of circadian gene oscillation in mouse aorta. Circulation. 2005; 112: 2716 -2724. PMID: 16230482 [PubMed] .
- 77. Peirson SN, Butler JN, Duffield GE, Takher S, Sharma P, Foster RG. Comparison of clock gene expression in SCN, retina, heart, and liver of mice. Biochem Biophys Res Commun. 2006; 351: 800 -807. [PubMed] .
- 78. Storch KF, Paz C, Signorovitch J, Raviola E, Pawlyk B, Li T, Weitz CJ. Intrinsic circadian clock of the mammalian retina: Importance for retinal processing of visual information. Cell. 2007; 130: 730 -741. PMID: 17719549 [PubMed] .
- 79. Hoogerwerf WA, Sinha M, Conesa A, Luxon BA, Shahinian VB, Cornelissen G, Halberg F, Bostwick J, Timm J, Cassone VM. Transcriptional profiling of mRNA expression in the mouse distal colon. Gastroenterology. 2008; 135: 2019 -2029. [PubMed] .
- 80. Yan J, Wang H, Liu Y, Shao C. Analysis of gene regulatory networks in the mammalian circadian rhythm. PLoS Comput Biol. 2008; 4: e1000193 [PubMed] .
- 81. Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, Hogenesch JB. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009; 5: e1000442 [PubMed] .
- 82. Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A. 2008; 105: 15172 -15177. [PubMed] .
- 83. Anea CB, Zhang M, Stepp DW, Simkins GB, Reed G, Fulton DJ, Rudic RD. Vascular disease in mice with a dysfunctional circadian clock. Circulation. 2009; 119: 1510 -1517. [PubMed] .
- 84. Hahn RA. Profound bilateral blindness and the incidence of breast cancer. Epidemiology. 1991; 2: 208 -10. [PubMed] .
- 85. Feychting M, Osterlund B, Ahlbom A. Reduced cancer incidence among the blind. Epidemiology. 1998; 9: 490 -494. [PubMed] .
- 86. Davis S, Mirick DK, Stevens RG. Night shift work, light at night, and risk of breast cancer. J Natl Cancer Inst. 2001; 93: 1557 -1562. [PubMed] .
- 87. Stevens RG and Rea MS. Light in the built environment: potential role of circadian disruption in endocrine disruption and breast cancer. Cancer Causes and Control. 2001; 12: 279 -87. [PubMed] .
- 88. Pukkala E, Ojamo M, Rudanko SL, Stevens RG, Verkasalo PK. Does incidence of breast cancer and prostate cancer decrease with increasing degree of visual impairment. Cancer Causes Control. 2006; 17: 573 -6. [PubMed] .
- 89. Stevens RG. Circadian disruption and breast cancer: from melatonin to clock genes. Epidemiology. 2005; 16: 254 -8. [PubMed] .
- 90. Flynn-Evans EE, Stevens RG, Tabandeh H, Schernhammer ES, Lockley SW. Total visual blindness is protective against breast cancer. Cancer Causes Control. 2009; 20: 1753 -6. [PubMed] .
- 91. Kantermann T and Roenneberg T. Is light-at-night a health risk factor or a health risk predictor? Chronobiol Int. 2009; 26: 1069 -1074. [PubMed] .
- 92. Straif K, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Altieri A, Benbrahim-Tallaa L, Cogliano V. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007; 8: 1065 -1066. [PubMed] .
- 93. Hill SM, Frasch T, Xiang S, Yuan L, Duplessis T, Mao L. Molecular mechanisms of melatonin anticancer effects. Integr Cancer Ther. 2009; 8: 337 -346. [PubMed] .
- 94. Davidson AJ, Sellix MT, Daniel J, Yamazaki S, Menaker M, Block GD. Chronic jet-lag increases mortality in aged mice. Curr Biol. 2006; 16: R914 -6. [PubMed] .
- 95. Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL, Besing RC, Menaker M, Gewirtz AT, Davidson AJ. Dysregulation of inflammatory responses by chronic circadian disruption. J Immunol. 2010; 185: 5796 -5805. [PubMed] .
- 96. Filipski E, Subramanian P, Carriere J, Guettier C, Barbason H, Levi F. Circadian disruption accelerates liver carcinogenesis in mice. Mutat Res. 2009; 680: 95 -105. [PubMed] .
- 97. Filipski E and Levi F. Circadian disruption in experimental cancer processes. Integr Cancer Ther. 2009; 8: 298 -302. [PubMed] .
- 98. Yasuniwa Y, Izumi H, Wang KY, Shimajiri S, Sasaguri Y, Kawai K, Kasai H, Shimada T, Miyake K, Kashiwagi E, Hirano G, Kidani A, Akiyama M, et al. Circadian disruption accelerates tumor growth and angio/stromagenesis through a wnt signaling pathway. PLoS One. 2010; 5: e15330 [PubMed] .
- 99. Preuss F, Tang Y, Laposky AD, Arble D, Keshavarzian A, Turek FW. Adverse effects of chronic circadian desynchronization in animals in a ‘challenging’ environment. Am J Physiol Regul Integr Comp Physiol. 2008; 295: R2034 -40. [PubMed] .
- 100. Roseboom PH, Namboodiri MA, Zimonjic DB, Popescu NC, Rodriguez IR, Gastel JA, Klein DC. Natural melatonin ‘knockdown’ in C57BL/6J mice: rare mechanism truncates serotonin N-acetyltransferase. Mol Brain Res. 1998; 63: 189 -197. [PubMed] .
- 101. Martino TA, Oudit GY, Herzenberg AM, Tata N, Koletar MM, Kabir GM, Belsham DD, Backx PH, Ralph MR, Sole MJ. Circadian rhythm disorganization produces profound cardiovascular and renal disease in hamsters. Am J Physiol Regul Integr Comp Physiol. 2008; 294: R1675 -83. [PubMed] .
- 102. Martino TA, Tata N, Belsham DD, Chalmers J, Straume M, Lee P, Pribiag H, Khaper N, Liu PP, Dawood F, Backx PH, Ralph MR, Sole MJ. Disturbed diurnal rhythm alters gene expression and exacerbates cardiovascular disease with rescue by resynchronization. Hypertension. 2007; 49: 1104 -1113. [PubMed] .
- 103. Scheer FA, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A. 2009; 106: 4453 -4458. [PubMed] .
- 104. Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U. Circadian gene expression in individual fibroblasts: Cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell. 2004; 119: 693 -705. [PubMed] .
- 105. Welsh DK, Yoo SH, Liu AC, Takahashi JS, Kay SA. Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr Biol. 2004; 14: 2289 -2295. [PubMed] .
- 106. Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000; 103: 1009 -1017. [PubMed] .
- 107. Kondratov RV, Vykhovanets O, Kondratova AA, Antoch MP. Antioxidant N–acetyl–L–cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging (Albany). 2009; 1: 979 -987. .
- 108. Bunger MK, Walisser JA, Sullivan R, Manley PA, Moran SM, Kalscheur VL, Colman RJ, Bradfield CA. Progressive arthropathy in mice with a targeted disruption of the Mop3/Bmal-1 locus. Genesis. 2005; 41: 122 -132. [PubMed] .
- 109. Alvarez JD, Hansen A, Ord T, Bebas P, Chappell PE, Giebultowicz JM, Williams C, Moss S, Sehgal A. The circadian clock protein BMAL1 is necessary for fertility and proper testosterone production in mice. J Biol Rhythms. 2008; 23: 26 -36. [PubMed] .
- 110. Boden MJ, Varcoe TJ, Voultsios A, Kennaway DJ. Reproductive biology of female Bmal1 null mice. Reproduction. 2010; 139: 1077 -1090. [PubMed] .
- 111. Sun Y, Yang Z, Niu Z, Wang W, Peng J, Li Q, Ma MY, Zhao Y. The mortality of MOP3 deficient mice with a systemic functional failure. J Biomed Sci. 2006; 13: 845 -51. [PubMed] .
- 112. McDearmon EL, Patel KN, Ko CH, Walisser JA, Schook AC, Chong JL, Wilsbacher LD, Song EJ, Hong HK, Bradfield CA, Takahashi JS. Dissecting the functions of the mammalian clock protein BMAL1 by tissue-specific rescue in mice. Science. 2006; 314: 1304 -1308. [PubMed] .
- 113. Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, Fitzgerald GA. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2004; 2: e377 [PubMed] .
- 114. Sadacca LA, Lamia KA, Delemos AS, Blum B, Weitz CJ. An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia. 2011; 54: 120 -124. [PubMed] .
- 115. Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, Lopez JP, Philipson LH, Bradfield CA, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010; 466: 627 -631. [PubMed] .
- 116. Yu E, Hussain S, Dallmann R, Russell S, Lian J, Weaver D. Age-dependent arthropathy in circadian mutant mice. J Bone Mineral Res. 2010; 25: Suppl 1 S97 (Abstract FR0154, Am Soc Bone Mineral Research) .
- 117. DeBruyne JP, Noton E, Lambert CM, Maywood ES, Weaver DR, Reppert SM. A clock shock: Mouse CLOCK is not required for circadian oscillator function. Neuron. 2006; 50: 465 -477. [PubMed] .
- 118. DeBruyne JP, Weaver DR, Reppert SM. Peripheral circadian oscillators require CLOCK. Curr Biol. 2007; 17: R538 -9. [PubMed] .
- 119. Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998; 280: 1564 -1569. [PubMed] .
- 120. King DP, Vitaterna MH, Chang AM, Dove WF, Pinto LH, Turek FW, Takahashi JS. The mouse Clock mutation behaves as an antimorph and maps within the W19H deletion, distal of kit. Genetics. 1997; 146: 1049 -1060. [PubMed] .
- 121. Dolatshad H, Campbell EA, O'Hara L, Maywood ES, Hastings MH, Johnson MH. Developmental and reproductive performance in circadian mutant mice. Hum Reprod. 2006; 21: 68 -79. [PubMed] .
- 122. Kennaway DJ, Boden MJ, Voultsios A. Reproductive performance in female clock Delta19 mutant mice. Reprod Fertil Dev. 2004; 16: 801 -810. [PubMed] .
- 123. Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994; 264: 719 -725. [PubMed] .
- 124. Yoo SH, Ko CH, Lowrey PL, Buhr ED, Song EJ, Chang S, Yoo OJ, Yamazaki S, Lee C, Takahashi JS. A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proc Natl Acad Sci U S A. 2005; 102: 2608 -2613. [PubMed] .
- 125. Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004; 101: 5339 -5346. [PubMed] .
- 126. Kondratov RV and Antoch MP. Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol. 2007; 17: 311 -317. [PubMed] .
- 127. Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998; 93: 929 -937. [PubMed] .
- 128. Bjarnason GA, Jordan RC, Wood PA, Li Q, Lincoln DW, Sothern RB, Hrushesky WJ, Ben-David Y. Circadian expression of clock genes in human oral mucosa and skin: Association with specific cell-cycle phases. Am J Pathol. 2001; 158: 1793 -1801. [PubMed] .
- 129. Brown SA, Fleury-Olela F, Nagoshi E, Hauser C, Juge C, Meier CA, Chicheportiche R, Dayer JM, Albrecht U, Schibler U. The period length of fibroblast circadian gene expression varies widely among human individuals. PLoS Biol. 2005; 3: e338 [PubMed] .
- 130. Pagani L, Semenova EA, Moriggi E, Revell VL, Hack LM, Lockley SW, Arendt J, Skene DJ, Meier F, Izakovic J, Wirz-Justice A, Cajochen C, Sergeeva OJ, et al. The physiological period length of the human circadian clock in vivo is directly proportional to period in human fibroblasts. PLoS One. 2010; 5: e13376 [PubMed] .
- 131. Sporl F, Schellenberg K, Blatt T, Wenck H, Wittern KP, Schrader A, Kramer A. A circadian clock in HaCaT keratinocytes. J Invest Dermatol. 2010; .
- 132. Zanello SB, Jackson DM, Holick MF. Expression of the circadian clock genes clock and period1 in human skin. J Invest Dermatol. 2000; 115: 757 -760. [PubMed] .
- 133. Tanioka M, Yamada H, Doi M, Bando H, Yamaguchi Y, Nishigori C, Okamura H. Molecular clocks in mouse skin. J Invest Dermatol. 2009; 129: 1225 -1231. [PubMed] .
- 134. Geyfman M and Andersen B. Clock genes, hair growth and aging. Aging (Albany NY). 2010; 2: 122 -128. [PubMed] .
- 135. Lin KK, Kumar V, Geyfman M, Chudova D, Ihler AT, Smyth P, Paus R, Takahashi JS, Andersen B. Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet. 2009; 5: e1000573 [PubMed] .
- 136. Reiter RJ, Paredes SD, Korkmaz A, Manchester LC, Tan DX. Melatonin in relation to the ‘strong’ and ‘weak’ versions of the free radical theory of aging. Adv Med Sci. 2008; 53: 119 -129. [PubMed] .
- 137. Siu AW, Maldonado M, Sanchez-Hidalgo M, Tan DX, Reiter RJ. Protective effects of melatonin in experimental free radical-related ocular diseases. J Pineal Res. 2006; 40: 101 -109. [PubMed] .
- 138. Kennaway DJ, Owens JA, Voultsios A, Varcoe TJ. Functional central rhythmicity and light entrainment, but not liver and muscle rhythmicity, are Clock independent. Am J Physiol Regul Integr Comp Physiol. 2006; 291: R1172 -80. [PubMed] .
- 139. Bertolucci C, Cavallari N, Colognesi I, Aguzzi J, Chen Z, Caruso P, Foa A, Tosini G, Bernardi F, Pinotti M. Evidence for an overlapping role of CLOCK and NPAS2 transcription factors in liver circadian oscillators. Mol Cell Biol. 2008; 28: 3070 -3075. [PubMed] .
- 140. Hong HK, Chong JL, Song W, Song EJ, Jyawook AA, Schook AC, Ko CH, Takahashi JS. Inducible and reversible Clock gene expression in brain using the tTA system for the study of circadian behavior. PLoS Genet. 2007; 3: e33 [PubMed] .
- 141. Spoelstra K, Oklejewicz M, Daan S. Restoration of self-sustained circadian rhythmicity by the mutant Clock allele in mice in constant illumination. J Biol Rhythms. 2002; 17: 520 -525. [PubMed] .
- 142. Kennaway DJ, Voultsios A, Varcoe TJ, Moyer RW. Melatonin and activity rhythm responses to light pulses in mice with the Clock mutation. Am J Physiol Regul Integr Comp Physiol. 2003; 284: R1231 -40. [PubMed] .
- 143. Ochi M, Sono S, Sei H, Oishi K, Kobayashi H, Morita Y, Ishida N. Sex difference in circadian period of body temperature in Clock mutant mice with Jcl/ICR background. Neurosci Lett. 2003; 347: 163 -166. [PubMed] .
- 144. Shimomura K, Lowrey PL, Vitaterna MH, Buhr ED, Kumar V, Hanna P, Omura C, Izumo M, Low SS, Barrett RK, LaRue SI, Green CB, Takahashi JS. Genetic suppression of the circadian Clock mutation by the melatonin biosynthesis pathway. Proc Natl Acad Sci U S A. 2010; 107: 8399 -8403. [PubMed] .
- 145. Dudley CA, Erbel-Sieler C, Estill SJ, Reick M, Franken P, Pitts S, McKnight SL. Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science. 2003; 301: 379 -383. [PubMed] .
- 146. Liu AC, Welsh DK, Ko CH, Tran HG, Zhang EE, Priest AA, Buhr ED, Singer O, Meeker K, Verma IM, Doyle FJ 3rd, Takahashi JS, Kay SA. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell. 2007; 129: 605 -616. [PubMed] .
- 147. Ko CH, Yamada YR, Welsh DK, Buhr ED, Liu AC, Zhang EE, Ralph MR, Kay SA, Forger DB, Takahashi JS. Emergence of noise-induced oscillations in the central circadian pacemaker. PLoS Biol. 2010; 8: e1000513 [PubMed] .
- 148. Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005; 308: 1043 -1045. [PubMed] .
- 149. Wu X, Wiater MF, Ritter S. NPAS2 deletion impairs responses to restricted feeding but not to metabolic challenges. Physiol Behav. 2010; 99: 466 -471. [PubMed] .
- 150. Herzog ED, Grace MS, Harrer C, Williamson J, Shinohara K, Block GD. The role of clock in the developmental expression of neuropeptides in the suprachiasmatic nucleus. J Comp Neurol. 2000; 424: 86 -98. [PubMed] .
- 151. Chappell PE, White RS, Mellon PL. Circadian gene expression regulates pulsatile gonadotropin-releasing hormone (GnRH) secretory patterns in the hypothalamic GnRH-secreting GT1-7 cell line. J Neurosci. 2003; 23: 11202 -11213. [PubMed] .
- 152. Miller BH, Olson SL, Levine JE, Turek FW, Horton TH, Takahashi JS. Vasopressin regulation of the proestrous luteinizing hormone surge in wild-type and Clock mutant mice. Biol Reprod. 2006; 75: 778 -784. [PubMed] .
- 153. Miller BH, Olson SL, Turek FW, Levine JE, Horton TH, Takahashi JS. Circadian Clock mutation disrupts estrous cyclicity and maintenance of pregnancy. Curr Biol. 2004; 14: 1367 -1373. [PubMed] .
- 154. Cermakian N, Monaco L, Pando MP, Dierich A, Sassone-Corsi P. Altered behavioral rhythms and clock gene expression in mice with a targeted mutation in the Period1 gene. EMBO J. 2001; 20: 3967 -3974. [PubMed] .
- 155. Pendergast JS, Friday RC, Yamazaki S. Endogenous rhythms in Period1 mutant suprachiasmatic nuclei in vitro do not represent circadian behavior. J Neurosci. 2009; 29: 14681 -14686. [PubMed] .
- 156. Zheng B, Larkin DW, Albrecht U, Sun ZS, Sage M, Eichele G, Lee CC, Bradley A. The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature. 1999; 400: 169 -173. [PubMed] .
- 157. Xu Y, Toh KL, Jones CR, Shin JY, Fu YH, Ptacek LJ. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell. 2007; 128: 59 -70. [PubMed] .
- 158. Pendergast JS, Friday RC, Yamazaki S. Distinct functions of Period2 and Period3 in the mouse circadian system revealed by in vitro analysis. PLoS One. 2010; 5: e8552 [PubMed] .
- 159. Ikeda H, Yong Q, Kurose T, Todo T, Mizunoya W, Fushiki T, Seino Y, Yamada Y. Clock gene defect disrupts light-dependency of autonomic nerve activity. Biochem Biophys Res Commun. 2007; 364: 457 -463. [PubMed] .
- 160. Dallmann R, Touma C, Palme R, Albrecht U, Steinlechner S. Impaired daily glucocorticoid rhythm in Per1brd mice. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2006; 192: 769 -775. [PubMed] .
- 161. Pilorz V and Steinlechner S. Low reproductive success in Per1 and Per2 mutant mouse females due to accelerated ageing? Reproduction. 2008; 135: 559 -568. [PubMed] .
- 162. Maronde E, Schilling AF, Seitz S, Schinke T, Schmutz I, van der Horst G, Amling M, Albrecht U. The clock genes period 2 and cryptochrome 2 differentially balance bone formation. PLoS One. 2010; 5: e11527 [PubMed] .