



Germline mutations in BRCA1 increase the risk of breast and ovarian cancer, but the specific pathways driving breast and ovarian cancer development in BRCA1 mutants are currently unclear [1]. Several studies have demonstrated that BRCA1 is required for cellular responses to DNA double strand breaks (DSBs) and homologous recombination (HR), although its exact role in these processes is unclear. BRCA1 contains an N-terminal RING domain and two C-terminal BRCT repeats. The BRCA1 RING domain imparts ubiquitin ligase activity to BRCA1 through interaction with its key binding partner BARD1 [2]. BRCT repeats contribute to transcriptional and DNA repair function of BRCA1, and cancer associated mutations that disrupt BRCT motif impair these activities [3].
BRCA1 forms at least 3 distinct complexes (BRCA1 A, BRCA1 B and BRCA1 C) through association of different adaptor proteins with BRCT motif. A-Complex function has been the subject of considerable research interest in the last several years, and is the focus of a study by Dever et al. in this issue of Aging [4] as well as other recent studies [5, 6]. The A-Complex consists of BRCA1 in association with the ubiquitin interacting motif (UIM) containing protein RAP80, the deubiquitinylating (DUB) enzymes BRCC36 and BRCC45, MERIT 40, and the adaptor protein Abraxas [7-10]. The A-Complex is thought to target BRCA1 to ionising radiation (IR) inducible foci through interaction of RAP80 with K63 poly-ubiquitin chains at sites of DSBs, in particular on H2AX [7, 11-13]. This RAP80 recruiting ubiquitinylation is performed by the E2 ubiquitin conjugase Ubc13 and the E3 ligase RNF8, which are targeted to breaks by MDC1 [14-16]. The critical role of ubiquitinylation as a recruitment signal at DNA damage sites is highlighted by the hypersensitivity of Ubc13 and RNF8-deficient cells to irradiation [14-17].
The new study by Dever et al. indicates that disruption of the BRCA1 complexes through mutation in BRCT motif causes genomic instability due to an increase in recombination [4]. Specifically, the authors explored the function of the BRCA1 BRCT mutant K1702M, in which BRCT mediated phosphoprotein interactions are disrupted. They found that expression of K1702M caused radiosensitivity compared to wild-type complemented BRCA1 deficient cells. Intra-chromosomal recombination is commonly assessed using a flow cytometry based DR-GFP recombination assay. Dever et al. used this method to determine that expression of the K1702M mutant was associated with elevated recombination compared to wild-type BRCA1, indicating that the K1702M mutant disrupts critical negative regulation of recombination [4]. It was unclear if this increased recombinational activity was due to the accumulation of cells in S/G2 [4]. The hyper-recombination of K1702M cells is in line with two other studies which found that transient depletion of non-BRCA1 members of the A-Complex caused an increase in recombination, using a similar DR-GFP approach [5, 6]. Interestingly, these studies found that siRNA depletion of BRCC36, one of the A-Complex deubiquitinylating enzymes, increased recombination activity to the same extent as RAP80 depletion[5, 6], seemingly contradicting earlier studies that reported reduced HR by depletion of the components of A-Complex [13, 18] and the reason for this lack of concordance is unclear. In support of the role of the BRCA1 BRCT domain and the A-Complex in negative regulation of recombination [4-6], is the finding that a balance between histone ubiquitinylation by RNF8/ Ubc13 and deubiquitinylation by BRCC36/RAP80 is critical for maintenance of genomic stability [19]. This supports a model where recruitment and subsequent deubiquitinylation activity of the A-Complex terminates DNA break associated ubiquitinylation. The importance of balanced poly-ubiquitin synthesis and removal at the break site is highlighted by regulation of FANCD2 [20]. Failure to ubiquitinate or deubiquitinate FANCD2 confers sensitivity to cross-linking agents and impairs recombination activity.
Recent work by Hu et al. showed that the hyper-recombination activity in RAP80 deficient cells is dependent on BRCA1, since double depletion of RAP80 and BRCA1 suppressed the increased HR observed in RAP80 deficient cells [6]. Dever et al. extended on this by showing that the hyper-recombination observed in BRCT-mutant expressing cells is dependent on BRCA1 ubiquitin ligase activity [4]. A BRCA1 RING domain mutation known to block BRCA1 ubiquitin ligase activity (I26A) was able to diminish increased recombination observed with the expression of the BRCT mutant K1702M [4]. Given the accumulation of K1702M mutant expressing cells in S/G2 phase of the cell cycle reported in this study [4], it would be interesting to know what effect the I26A/ K1702M double mutant has on cell cycle. Taken along with the hyper-recombination of BRCC36 depleted cells, these results support a model where the A-Complex negatively regulates the pro-recombination activity of an ubiquitinylation-dependant BRCA1 pathway.
Some questions raised from this conclusion are: What recombination mechanism is the A-Complex regulating? What effects does A-Complex disruption have on the structure of chromatin surrounding a break site? How does A-Complex recruitment and deubiquitinylase activity affect recruitment of other factors? The mechanism of hyper-recombination in A-Complex disrupted cells has been addressed by Hu et al. [6] and in the Dever et al. [4] study. They found that ablation of the BRCA1-Abraxas interaction promotes extensive resection, as visualized by an increase in single stranded DNA abundance and chromatin retention of RPA and RAD51[4, 6]. Elevated binding of RPA and RAD51 is indicative of more abundant or longer stretches of resected single stranded DNA. In fact Hu et al. demonstrated that a single I-SceI induced DSB is resected further in cells lacking RAP80, by mapping RPA and Rad51 binding around the break by chromatin-immunoprecipitation (ChIP)-PCR [6]. Dever et al. directly measured single stranded DNA by detecting BrdU incorporation in non-denaturing conditions [4]. Both studies showed that disruption of BRCT interactions causes an increase in the abundance of single stranded DNA [4, 6]. Thus the hyper-recombination of A-Complex disrupted cells observed in several studies is the result of excessive resection, possibly due to loss of A-Complex catalysed termination of ubiquitinylation (deubiquitinylation) at breaks [19] (Figure 1).
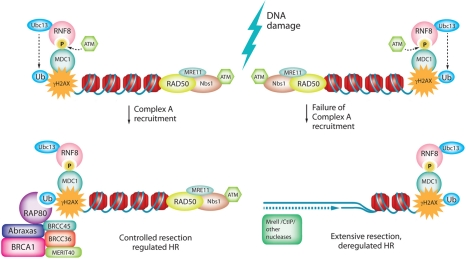
Figure 1. A-Complex regulates DSB resection
Taken together these recent studies provide firm evidence that the BRCA1 A-Complex fine-tunes the repair function of BRCA1 by repressing excessive resection in a BRCT dependent manner. Interesting areas for further study may include elucidating the role of post-translational modifications in A-Complex formation and regulation. It would also be interesting to examine the interaction profile of BRCT mutants to identify mediators of excessive recombination. The hyper-recombination observed in BRCC36 depleted cells and the decreased recombination observed in Usp1 (FANCD2 deubiquitinylase) deleted cells highlight a central role for different ubiquitin marks on chromatin and repair proteins. Identification of A-Complex deubiquitinylase targets and downstream effects would provide valuable insight into the mechanism of hyper-recombination in A-Complex defective cells.
Double strand breaks are detected by the sensor protein complex Mre11-Rad50-Nbs1 (MRN), which recruits and activates the kinase ATM. Activated ATM phosphorylates many DNA repair proteins including MDC1. ATM phosphorylated MDC1 recruits the RNF8-Ubc13 E3 ubiquitin ligase which synthesizes K63 linked polyubiquitin on histones H2A and H2AX. RAP80 targets the A-Complex to breaks via the interactions of its ubiquitin interacting motifs (UIMs) with K63 linked polyubiquitin. Ubiquitin-dependent A-Complex localization to chromatin and subsequent deubiquitinylation fine tunes resection, resulting in tightly controlled recombination. Failure of A-Complex recruitment due to mutation in the BRCT motif, or depletion of A-Complex components including BRCC36/45 (deubiquitinylases) would alter the chromatin ubiquitinylation pattern. Such persistence or aberration of chromatin marks would change the complement and activity of repair proteins at the break site, resulting in de-regulation of repair. We propose that deubiquitinylase activity of the A-Complex restricts the recruitment and activity of nucleases, so disruption of the A-Complex leads to extensive resection and uncontrolled recombination.
Acknowledgments
We would like to thank Madeline Kersting for her assistance with figure preparation.
References
- 1. Elledge SJ and Amon A. The BRCA1 suppressor hypothesis: an explanation for the tissue-specific tumor development in BRCA1 patients. Cancer Cell. 2002; 1: 129 -132. [PubMed] .
- 2. Wu-Baer F, Lagrazon K, Yuan W, Baer R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J Biol Chem. 2003; 278: 34743 -34746. [PubMed] .
- 3. Scully R and Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000; 408: 429 -432. [PubMed] .
- 4. Dever SM, Golding SE, Rosenberg E, Adams BR, Idowu MO, Quillin JM, Valerie M, Xu B, Povirk LF, Valerie K. Mutations in the BRCT Binding Site of BRCA1 Result in Hyper-recombination. Ageing. 2011. current issue. .
- 5. Coleman KA and Greenberg RA. The BRCA1-RAP80 Complex Regulates DNA Repair Mechanism Utilization by Restricting End Resection. J Biol Chem. 2011; 286: 13669 -13680. [PubMed] .
- 6. Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011; 25: 685 -700. [PubMed] .
- 7. Wang B and Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci U S A. 2007; 104: 20759 -20763. [PubMed] .
- 8. Dong Y, Hakimi MA, Chen X, Kumaraswamy E, Cooch NS, Godwin AK, Shiekhattar R. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol Cell. 2003; 12: 1087 -1099. [PubMed] .
- 9. Shao G, Patterson-Fortin J, Messick TE, Feng D, Shanbhag N, Wang Y, Greenberg RA. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009; 23: 740 -754. [PubMed] .
- 10. Wang B, Hurov K, Hofmann K, Elledge SJ. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009; 23: 729 -739. [PubMed] .
- 11. Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007; 316: 1202 -1205. [PubMed] .
- 12. Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007; 316: 1198 -1202. [PubMed] .
- 13. Yan J, Kim YS, Yang XP, Li LP, Liao G, Xia F, Jetten AM. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 2007; 67: 6647 -6656. [PubMed] .
- 14. Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007; 131: 901 -914. [PubMed] .
- 15. Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007; 318: 1637 -1640. [PubMed] .
- 16. Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007; 131: 887 -900. [PubMed] .
- 17. Zhao GY, Sonoda E, Barber LJ, Oka H, Murakawa Y, Yamada K, Ikura T, Wang X, Kobayashi M, Yamamoto K, et al. A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol Cell. 2007; 25: 663 -675. [PubMed] .
- 18. Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007; 316: 1194 -1198. [PubMed] .
- 19. Shao G, Lilli DR, Patterson-Fortin J, Coleman KA, Morrissey DE, Greenberg RA. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc Natl Acad Sci U S A. 2009; 106: 3166 -3171. [PubMed] .
- 20. Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D'Andrea AD. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell. 2009; 16: 314 -320. [PubMed] .