



Introduction
Alzheimer's disease (AD) is the most common form of dementia in the ageing population and affects millions of people worldwide [1]. At the neuropathological level, AD is characterized by neuronal cell loss and the combined presence of two lesions in the brain - extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) [2]. The extracellular deposits contain aggregated Aβ peptides [3], while intraneuronal tangles are aggregates of hyper-phosphorylated forms of the neurofilament-associated protein tau [4]. Evidence suggests that the pathogenesis of AD involves deleterious neurotoxic effects of both types of aggregates [5;6]. However, numerous studies strongly support a critical role of Aβ aggregates in the initiation phase of AD pathogenesis, while tau might mediate toxicity and impairment of neuronal function [5-9].
Aβ is a proteolytically processed fragment of the amyloid precursor protein (APP) [10;11]. It occurs in different length variants with peptides of 40 amino acid residues (Aβ40) and 42 amino acid residues (Aβ42) being the most prevalent. The longer Aβ42 variant has a much higher propensity to form aggregates. Genetic studies identified mutations in three genes that cause familial forms of AD (FAD): APP, presenilin-1 (PS1), and presenilin-2 (PS2) [12]. Mutations in each of these genes result in elevated levels of Aβ production and/or promote its aggregation. This genetic correlation strongly favours the key role of Aβ in AD. However, mutations in APP and PS are very rare, and the causes of the much more common late onset forms of AD (LOAD) are largely unidentified. In line with a significant role of Aβ in pathogenesis, recent data show that various post-translational modifications of Aβ promote its aggregation and therefore could play important roles in the initiation of LOAD.
Generation of Aβ by proteolytic processing of APP and effects of AD associated mutations
APP is a type I membrane protein and ubiquitously expressed in most cell types. Alternative mRNA splicing leads to several cell type and development-specific isoforms [2]. In addition, two homologous APP-like proteins (APLPs) have also been identified, that together form a small protein family with important physiological functions in perinatal and postnatal development and cell communication [13]. However, APLPs do not contain the Aβ sequence and thus APP is the sole source of Aβ peptides in the brain [14].
Aβ is produced during normal cellular metabolism and secreted to the extracellular milieu of the human brain and also found in cerebrospinal fluid (CSF) [15;16]. The presence of Aβ in the CSF of nondemented individuals and in the media from neuronal cell cultures during normal metabolism could indicate a physiological role of Aβ in the central nervous system [17]. Suggested physiological function of Aβ includes ion channel modulation [18], kinase activation [19], regulation of cholesterol transport [20], protection against metal-induced oxidative damage [21], learning and memory [22] and transcriptional regulation of AD-associated genes [23].
The generation of Aβ is initially starts with a cleavage of APP by β-secretase at the N-terminus of the Aβ domain (Figure 1A). This cleavage results in the shedding of the APP ectodomain and the generation of a membrane bound carboxyl (C)-terminal fragment (CTF-β). Subsequently, γ-secretase mediates the apparently intramembranous cleavage of CTF-β resulting in the liberation of Aβ into conditioned media of cultured cells or extracellular fluids of the brain or the periphery [2;11]. Alternatively, APP can also be cleaved in a non-amyloidogenic pathway that involves initial cleavage by α-secretase within the Aβ domain thereby precluding the subsequent generation of Aβ peptides (Figure 1A) [24].
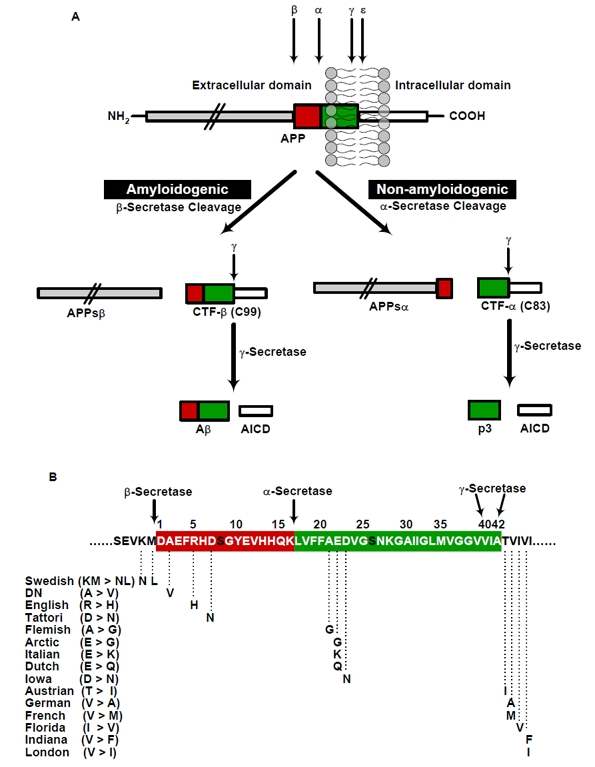
Figure 1. Schematic representation of generation of Aβ by proteolytic processing of APP and the familial AD causing APP mutations (A) Two pathways (β/γ and α/γ) of APP proteolysis. APP can be cleaved by either β- or α-secretase, which is then followed by γ-secretase cleavage results in the generation of either the p3-fragment (non-amyloidogenic) or an Aβ (amyloigenic pathway). The designation of secretases, substrates and products are depicted, (B) Representation of APP familial AD causing mutations that are identified around N- and C-terminal and in the middle region of Aβ. The amino acid residues are numbered according to Aβ sequence. The swedish mutation (KM>NL) at N-terminus of Aβ̣ near to β-secretase cleavage site increases the total production of Aβ, whereas the mutations C-terminus of Aβ results in increased production of Aβ42 by altering γ-secretase activity. The mutations in the middle region of Aβ might decrease the α-secretory cleavage, facilitate the amyloidogenic processing, promote the Aβ production and/or increases the propensity of Aβ aggregation or stabilizes the Aβ against clearance by different proteases.
The mutations within APP that causes early onset AD (EOAD), are all located within or close to the Aβ domain. Notably, a double mutation in APP at the cleavage site for β-secretase that cause EOAD increases the β-secretory cleavage resulting in an overall higher production of Aβ peptides (see Swedish mutation, Figure 1B) [25]. Additional EOAD-associated mutations located close to the cleavage site for γ-secretase at c-terminal of Aβ also alter the proteolytic processing of APP (Figure 1B). These mutations increase the ratio of Aβ42/40 peptides thereby promoting the relative production of Aβ variants with higher propensity to aggregate [26]. Mutations found in the middle of the Aβ domain might exert different effects (Figure 1B), (1) they might decrease the α-secretory cleavage of APP thereby facilitating amyloidogenic processing of APP [27], (2) these mutations could also increase the aggregation [28], (3) and/or alter the degradation by different proteases [29].
Beside the APP gene, two additional genes have been identified to contain mutations that lead to EOAD [30]. Both genes encode highly homologous PS proteins that are critical components of the γ-secretase complex, which includes three additional proteins such as nicastrin, APH-1 (anterior pharynx-defective 1), and PEN-2 (presenilin enhancer 2) to exert γ-secretase activity in cells [31]. The mutations in PS1 or PS2 also alter γ-secretase activity and/or cleavage specificity, resulting in higher ratios of Aβ42/40 [31]. Together, all mutations in the three genes known to be associated with EOAD affect the generation and/or aggregation of Aβ [25;27]. However, as mentioned before such mutations are very rare and mechanisms that increase the aggregation and accumulation of Aβ and cause the much more common sporadic forms of AD (>95% of all cases), are largely unknown. According to the ‘amyloid hypothesis‘, accumulation of Aβ in the brain is the primary influence driving AD pathogenesis. The rest of the pathogenic events, including impaired synaptic function and cell communication [7;32;33], activation of microglia and astrocytes [34;35], neuronal ionic homeostasis and oxidative injury [36], mitochondrial dysfunction [37], altered kinase/phosphatases activities leading to formation of neurofibrillary tangles containing tau protein, is proposed to result from an imbalance between Aβ production and Aβ clearance [38].
Aβ aggregation – routes to neurotoxic assemblies
Amyloid formation in AD is conceptualized as a complex process of protein aggregation, involving the misfolding of Aβ into soluble and insoluble assemblies [39]. Monomeric Aβ is mainly composed of α-helical and/or unordered structure, whereas the misfolded polymers are rich in β-sheet conformation. The conformational changes leading to the formation of extended β-sheets promotes homophilic interactions and eventually leads to Aβ oligomer formation. Kinetic studies have suggested that misfolding of monomeric Aβ precedes formation of oligomers, which then serve as seeds/nuclei for accelerated fibril growth (Figure 2) [40].
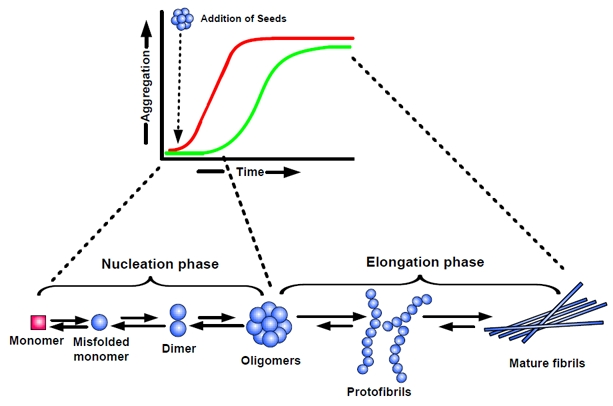
Figure 2. Nucleation-dependent polymerization model of amyloid aggregation Amyloid formation consists of two phases: (i) a nucleation phase/lag phase, in which monomers undergo conformational change/misfolding and associate to form oligomeric nuclei, and (ii) a elongation phase/growth phase, in which the nuclei rapidly grow by further addition of monomers and form larger polymers/fibrils until saturation. The ‘nucleation phase‘, is thermodynamically unfavourable and occurs gradually, whereas ‘elongation phase’, is much more favourable process and proceeds quickly. Thus, kinetics of amyloid formation is well represented by a sigmoidal curve with a lag phase followed by rapid growth phase (green curve). The rate limiting step in the process is the formation of nuclei/seeds to promote aggregation. Thus, amyloid formation can be substantially speedup by the addition of preformed seeds (nuclei). The addition of seeds reduces the lag time and induces faster aggregate formation (red curve).
A widely accepted concept for the formation of amyloid fibrils is the nucleation-dependent polymerization model [41-43], which separates the fibrillization process into a nucleation phase and an elongation phase. Nucleation requires the self-association of soluble monomers, which is thermodynamically unfavourable and so occurs slowly. In the nucleation phase, monomers undergo conformational changes and self-associate to form oligomeric nuclei that are rich in β-sheets. Once the nucleus is formed, assembly of larger aggregates and fibril elongation, a much more favourable process and proceeds rapidly. As a result, the kinetics of amyloid fibril formation is well represented by a sigmoidal shape with a nucleation phase/lag phase followed by a rapid growth phase, followed by a saturation phase (Figure 2; green curve). The lag phase is determined by the critical concentration of nuclei, which represent seeds for further growth of the polymers finally resulting in mature fibrils. Accordingly, the lag phase of aggregation can be shortened by addition of preformed seeds (Figure 2; red curve).
In a landmark discovery, Pike et al., [44], established that innocuous monomers of Aβ become neurotoxic upon aggregation. It was further shown that toxicity of Aβ involved self-association of monomers into oligomers and higher aggregated forms [45]. This is further supported by in vitro [46-48], and in vivo studies showing that oligomeric and pre-fibrillar Aβ assemblies are potent neurotoxins [5;49;50]. A correlation between soluble oligomeric Aβ levels and the extent of synaptic loss and severity of cognitive impairment further corroborate the findings [7;32]. Thus, neurotoxicity appears to require toxic oligomeric assemblies of Aβ. The formation of such neurotoxic assemblies in the brain generated due to higher production and/or decreased clearance of Aβ [51;52].
Effect of post-translational modification on aggregates formation, toxicity and clearance
Amyloid plaques in the human AD brain are known to contain a heterogeneous mixture of Aβ peptides [53]. In addition to main Aβ species (Aβ40 and Aβ42), a variety of post-translationally modified variants have been identified [54], including truncation [55-58], racemization [59;60], isomerization [61;62], pyroglutamination [63;64], metal induced oxidation [65] and phosphorylation [66-68].
The N-terminal truncated variants of Aβ beginning at amino acid 3, 11 and 25 are present in senile plaques and vascular amyloid deposits [56;57;57;69-71]. The truncated Aβ25-35 is shown to favour aggregation in vitro [72]. Due to potential toxic effects of truncated Aβ25-35, it has been frequently used for aggregation or toxicity studies [73]. Racemization of Aβ at Asp7, Asp23 and Ser26 was reported in the human brain and aggregation properties of Aβ were influenced by the position of the racemized residue [59;60]. Isomerization of aspartate residues at position 1, 7 and 23 of Aβ results in structural transition of Aβ and also shown to occur in vivo [62]. Isomerization of Aβ promotes fibril formation in vitro and resistance to proteolytic degradation [61]. In addition Aβ can undergo pyroglutamination also resulting in faster aggregation [74;75].
Thus, post-translational modifications of Aβ could promote oligomer and aggregate formation, thereby also reducing the degradation by a variety of proteases [76-79]. Modified Aβ peptides show enhanced cytotoxicity as compared to non-modified peptides [73], and serve as seeding species for Aβ aggregate formation in vivo [66;74;78]. These post-translationally modified Aβ variants appear to be present at an early stages of the disease [58;66;71;74].
Extracellular phosphorylation
Phosphorylation is an important reversible post-translational modification that regulates the structural and functional properties of proteins in health and disease [80]. Phosphorylation is a key step in the regulation of protein activity, cell cycle control, gene regulation, learning and memory [81]. In addition to intracellular protein kinases (PKs), extracellular PK activities have also been described [82]. These extracellular kinases phosphorylate cell-surface proteins and soluble extracellular substrates, and thus could affect many physiological processes involving cell-cell contacts, cellular differentiation and proliferation, ion transport [82]. Depending on the localization, these PKs are differentiated as ecto-PKs and exo-PKs. Ecto-PKs are localized at the external surface of the plasma membrane (membrane bound) where they exert their catalytic activity [83-86]. Exo-PKs are secreted/shedded to the extracellular milieu [87;88]. Ecto- and Exo-PKs can phosphorylate extracellular membrane bound proteins and soluble proteins Both Ecto- and Exo-PKs use extracellular ATP as co-substrate, which can be released by intact cells [89;90]. Extracellular ATP plays physiological roles in neurite outgrowth, neurotransmission and glial communication [91]. The release of extracellular ATP is mediated by metabotropic (P2Y) and ionotropic (P2X) receptors, both are widely expressed in the nervous system [92]. In the brain, extracellular ATP is present in low nanomolar concentrations. However, the local ATP concentration can increase upon certain stimuli, including synaptic activation [89;93], inflammation [94] and ischaemia in vivo [95]. Therefore, extracellular phosphorylation is likely to play a role in normal as well as pathological processes in the brain.
Phosphorylation of Aβ
A variety of AD associated proteins including APP [96-98], BACE [99;100], PS [101;102] and tau [103;104], are shown to be phosphorylated. Phosphorylation of these proteins affects subcellular trafficking, interaction with adapter proteins, signal transduction cascades, APP processing, Aβ generation and tangle formation. In AD, tau is shown to be abnormally hyperphosphorylated at several Ser/Thr residues. Hyperphosphorylation and subsequent accumulation of neurofilament subunits is a typical feature of the AD brain [105;106]. However, the pathophysiological relevance of tau phosphorylation is still under debate.
In silico analysis revealed that Aβ contain potential phosphorylation sites at serine residue at 8th and 26th position and tyrosine residue at 10th position. Aβ can undergo phosphorylation by protein kinase A and cdc 2 in vitro [68], as well as by cultured cells and in human CSF (Kumar, 2009; URN: urn:nbn:de:hbz:5N-18193).
We recently showed that Aβ is phosphorylated at serine-8 by extracellular protein kinase A. Phosphorylation of Aβ promoted the formation of toxic aggregates [66]. The formation of small soluble oligomers is associated with the conformational transition of Aβ from α-helical and random coiled state to a β-sheet structure, as demonstrated by circular dichroism. Phosphorylation-state specific antibodies were used in western-blotting and immunohistochemistry to demonstrate the occurrence of phosphorylated Aβ in murine AD models and AD patient's brain tissue. Notably, these antibodies further confirmed that phosphorylation occurs at free extracellular Aβ rather than at the full-length APP or β-CTF, the precursors of Aβ peptide. Phosphorylated Aβ co-localized with non-phosphorylated Aβ in extracellular plaques [66]. Interestingly, phosphorylated Aβ appeared to be concentrated in the centre of individual plaques and was detected as early as at 2 months of age in APP transgenic mice, and then accumulated with aging. The detection of phosphorylated Aβ in oligomeric assemblies in mouse brain homogenates suggested that phosphorylation also increases aggregation of Aβ in vivo. Therefore, we hypothesize that phosphorylation of Aβ might act as a conformational switch, thereby promoting the formation of aggregates.
To test the effect of Aβ phosphorylation on toxicity in vivo, transgenic Drosophila models were employed. Since Drosophila allows the selective expression of Aβ independent of its precursor APP [107;108], transgenic Drosophila flies expressing either the wild type Aβ (AβWT) or pseudophosphorylated mutant (AβS8D) were generated. When expressing AβWT and AβS8D mutant in photoreceptor cells in Drosophila eyes, the pseudophosphorylated AβS8D variant showed significant cell degeneration compared to AβWT, demonstrating increased toxicity of pseudophosphorylated Aβ. Notably, pseudophosphorylated AβS8D also accumulated to much higher levels in aged flies than AβWT, strongly indicating increased aggregation. In addition, transgene expression in the fly brain showed stronger age-dependent accumulation of pseudophoshporylated Aβ peptides as compared to AβWT. The increased toxicity of pseudophosphorylated Aβ was revealed by altered climbing behaviour upon aging. This progressive age-dependent phenotype, correlates with Aβ peptide accumulation, indicating that pseudophosphorylated Aβ can mimic the effect of phosphorylation on Aβ aggregation in vivo [66].
The Aβ plaque formation could be induced by inoculation of amyloid containing brain homogenates from human or transgenic mouse into brains of monkeys or APP transgenic mice, suggesting the occurrence of nucleation-dependent fibrillization in vivo [109;110]. As phosphorylation of Aβ promotes oligomer formation, phosphorylated Aβ oligomers could serve as seeds or nuclei that increase the rate of aggregation. In agreement with this hypothesis, the nuclei of phosphorylated Aβ were capable to promote aggregation of non-phosphorylated Aβ in vitro [66].
Several proteases or peptidases have been reported that are able to cleave Aβ and thereby contribute to efficient removal of Aβ in the brain [52;111]. It will therefore also be interesting to assess the effect of phosphorylation on protease dependent degradation of Aβ.
Conclusion
Increasing evidence suggests that phosphorylation of proteins involved in several neurodegenerative diseases and plays a serious role during the pathogenesis [67;112;113]. The role of phosphorylation in modulating the aggregation and fibrillogenesis of tau in AD and α-synuclein in Parkinson's disease (PD) is currently a subject of intense investigation [103;114;115]. Our studies provide evidence that Aβ can undergo phosphorylation. Phosphorylation promotes conformational transition and formation of toxic aggregates. Further, phosphorylated Aβ aggregates could serve as endogenous seeds triggering further aggregation of soluble, extracellular Aβ into plaques in the brain. Phosphorylation stabilizes the Aβ against degradation by various proteases in vitro and in cell cultures (Kumar et al., Unpublished data). The stabilization of Aβ by phosphorylation might play a crucial role in AD pathogenesis, because it would eventually result in increased concentrations of this peptide in the brain. Therefore, inhibition of extracellular kinases or stimulation of Aβ dephosphorylation could be pursued as valuable targets to prevent or slow down the progression of AD. Further, the detection of phosphorylated Aβ in biological fluids could also be explored for evaluation as biomarkers. Together, phosphorylation of Aβ might have very important implications for AD pathogenesis and offer novel therapeutic avenues.
Acknowledgments
We thank Dr. Peter Breuer and Dr. Patrick Wunderlich for critically reading the manuscript. Although we have made a thorough and extensive search of the literature, we apologize to our colleagues if we mistakenly excluded their studies in our reference list. Work in the laboratory was supported by Deutsche Forschungsgemeinschaft (DFG) grant (WA1477/6, SFB645, KFo177).
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Thies W and Bleiler L. 2011 Alzheimer's disease facts and figures. Alzheimers Dement. 2011; 7: 208 -244. [PubMed] .
- 2. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001; 81: 741 -766. [PubMed] .
- 3. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985; 82: 4245 -4249. [PubMed] .
- 4. Querfurth HW and LaFerla FM. Alzheimer's disease. N Engl J Med. 2010; 362: 329 -344. [PubMed] .
- 5. Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008; 192: 106 -113. [PubMed] .
- 6. LaFerla FM. Pathways linking Abeta and tau pathologies. Biochem Soc Trans. 2010; 38: 993 -995. [PubMed] .
- 7. Haass C and Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007; 8: 101 -112. [PubMed] .
- 8. Trojanowski JQ, Shin RW, Schmidt ML, Lee VM. Relationship between plaques, tangles, and dystrophic processes in Alzheimer's disease. Neurobiol Aging. 1995; 16: 335 -340. [PubMed] .
- 9. Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010; 330: 198 [PubMed] .
- 10. Haass C and De Strooper B. The presenilins in Alzheimer's disease—proteolysis holds the key. Science. 1999; 286: 916 -919. [PubMed] .
- 11. Walter J, Kaether C, Steiner H, Haass C. The cell biology of Alzheimer's disease: uncovering the secrets of secretases. Curr Opin Neurobiol. 2001; 11: 585 -590. [PubMed] .
- 12. Goate AM. Molecular genetics of Alzheimer's disease. Geriatrics. 1997; 52: Suppl 2 S9 -12. [PubMed] .
- 13. Anliker B and Muller U. The functions of mammalian amyloid precursor protein and related amyloid precursor-like proteins. Neurodegener Dis. 2006; 3: 239 -246. [PubMed] .
- 14. Walsh DM, Minogue AM, Sala FC, Fadeeva JV, Wasco W, Selkoe DJ. The APP family of proteins: similarities and differences. Biochem Soc Trans. 2007; 35: 416 -420. [PubMed] .
- 15. Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992; 359: 325 -327. [PubMed] .
- 16. Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992; 359: 322 -325. [PubMed] .
- 17. Pearson HA and Peers C. Physiological roles for amyloid beta peptides. J Physiol. 2006; 575: 5 -10. [PubMed] .
- 18. Kerrigan TL, Atkinson L, Peers C, Pearson HA. Modulation of ‘A’-type K+ current by rodent and human forms of amyloid beta protein. Neuroreport. 2008; 19: 839 -843. [PubMed] .
- 19. Tabaton M, Zhu X, Perry G, Smith MA, Giliberto L. Signaling effect of amyloid-beta(42) on the processing of AbetaPP. Exp Neurol. 2010; 221: 18 -25. [PubMed] .
- 20. Yao ZX and Papadopoulos V. Function of beta-amyloid in cholesterol transport: a lead to neurotoxicity. FASEB J. 2002; 16: 1677 -1679. [PubMed] .
- 21. Zou K, Gong JS, Yanagisawa K, Michikawa M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci. 2002; 22: 4833 -4841. [PubMed] .
- 22. Morley JE, Farr SA, Banks WA, Johnson SN, Yamada KA, Xu L. A physiological role for amyloid-beta protein:enhancement of learning and memory. J Alzheimers Dis. 2010; 19: 441 -449. [PubMed] .
- 23. Bailey JA, Maloney B, Ge YW, Lahiri DK. Functional activity of the novel Alzheimer's amyloid beta-peptide interacting domain (AbetaID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes and in amyloidogenesis. Gene. 2011; .
- 24. Kojro E and Fahrenholz F. The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem. 2005; 38: 105 -127. [PubMed] .
- 25. Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992; 360: 672 -674. [PubMed] .
- 26. Herl L, Thomas AV, Lill CM, Banks M, Deng A, Jones PB, Spoelgen R, Hyman BT, Berezovska O. Mutations in amyloid precursor protein affect its interactions with presenilin/gamma-secretase. Mol Cell Neurosci. 2009; 41: 166 -174. [PubMed] .
- 27. Haass C, Hung AY, Selkoe DJ, Teplow DB. Mutations associated with a locus for familial Alzheimer's disease result in alternative processing of amyloid beta-protein precursor. J Biol Chem. 1994; 269: 17741 -17748. [PubMed] .
- 28. Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003; 424: 805 -808. [PubMed] .
- 29. Betts V, Leissring MA, Dolios G, Wang R, Selkoe DJ, Walsh DM. Aggregation and catabolism of disease-associated intra-Abeta mutations: reduced proteolysis of AbetaA21G by neprilysin. Neurobiol Dis. 2008; 31: 442 -450. [PubMed] .
- 30. Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999; 13: 2801 -2810. [PubMed] .
- 31. De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007; 8: 141 -146. .
- 32. Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007; 27: 796 -807. [PubMed] .
- 33. Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004; 24: 10191 -10200. [PubMed] .
- 34. Choi SH and Bosetti F. Cyclooxygenase-1 null mice show reduced neuroinflammation in response to beta-amyloid. Aging (Albany NY). 2009; 1: 234 -244. [PubMed] .
- 35. Candelario-Jalil E. A role for cyclooxygenase-1 in beta-amyloid-induced neuroinflammation. Aging (Albany NY). 2009; 1: 350 -353. [PubMed] .
- 36. Pratico D and Delanty N. Oxidative injury in diseases of the central nervous system: focus on Alzheimer's disease. Am J Med. 2000; 109: 577 -585. [PubMed] .
- 37. Massaad CA, Pautler RG, Klann E. Mitochondrial superoxide: a key player in Alzheimer's disease. Aging (Albany NY). 2009; 1: 758 -761. [PubMed] .
- 38. Hardy J and Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002; 297: 353 -356. [PubMed] .
- 39. Yoshiike Y, Minai R, Matsuo Y, Chen YR, Kimura T, Takashima A. Amyloid oligomer conformation in a group of natively folded proteins. PLoS One. 2008; 3: e3235 [PubMed] .
- 40. Ni CL, Shi HP, Yu HM, Chang YC, Chen YR. Folding stability of amyloid-beta 40 monomer is an important determinant of the nucleation kinetics in fibrillization. FASEB J. 2011; 25: 1390 -1401. [PubMed] .
- 41. Jarrett JT and Lansbury PT Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993; 73: 1055 -1058. [PubMed] .
- 42. Harper JD and Lansbury PT Jr. Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997; 66: 385 -407. [PubMed] .
- 43. Naiki H and Gejyo F. Kinetic analysis of amyloid fibril formation. Methods Enzymol. 1999; 309: 305 -318. [PubMed] .
- 44. Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of beta-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991; 563: 311 -314. [PubMed] .
- 45. Lorenzo A and Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci U S A. 1994; 91: 12243 -12247. [PubMed] .
- 46. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998; 95: 6448 -6453. [PubMed] .
- 47. Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999; 19: 8876 -8884. [PubMed] .
- 48. Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006; 572: 477 -492. [PubMed] .
- 49. Walsh DM and Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007; 101: 1172 -1184. [PubMed] .
- 50. Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001; 24: 219 -224. [PubMed] .
- 51. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010; 330: 1774 [PubMed] .
- 52. Iwata N, Higuchi M, Saido TC. Metabolism of amyloid-beta peptide and Alzheimer's disease. Pharmacol Ther. 2005; 108: 129 -148. [PubMed] .
- 53. Walker LC, Rosen RF, LeVine H III. Diversity of Abeta deposits in the aged brain: a window on molecular heterogeneity? Rom J Morphol Embryol. 2008; 49: 5 -11. [PubMed] .
- 54. Kuo YM, Kokjohn TA, Beach TG, Sue LI, Brune D, Lopez JC, Kalback WM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Roher AE. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer's disease brains. J Biol Chem. 2001; 276: 12991 -12998. [PubMed] .
- 55. Saido TC, Yamao-Harigaya W, Iwatsubo T, Kawashima S. Amino- and carboxyl-terminal heterogeneity of beta-amyloid peptides deposited in human brain. Neurosci Lett. 1996; 215: 173 -176. [PubMed] .
- 56. Tekirian TL, Saido TC, Markesbery WR, Russell MJ, Wekstein DR, Patel E, Geddes JW. N-terminal heterogeneity of parenchymal and cerebrovascular Abeta deposits. J Neuropathol Exp Neurol. 1998; 57: 76 -94. [PubMed] .
- 57. Miravalle L, Calero M, Takao M, Roher AE, Ghetti B, Vidal R. Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry. 2005; 44: 10810 -10821. [PubMed] .
- 58. Hartig W, Goldhammer S, Bauer U, Wegner F, Wirths O, Bayer TA, Grosche J. Concomitant detection of beta-amyloid peptides with N-terminal truncation and different C-terminal endings in cortical plaques from cases with Alzheimer's disease, senile monkeys and triple transgenic mice. J Chem Neuroanat. 2010; 40: 82 -92. [PubMed] .
- 59. Mori H, Ishii K, Tomiyama T, Furiya Y, Sahara N, Asano S, Endo N, Shirasawa T, Takio K. Racemization: its biological significance on neuropathogenesis of Alzheimer's disease. Tohoku J Exp Med. 1994; 174: 251 -262. [PubMed] .
- 60. Tomiyama T, Asano S, Furiya Y, Shirasawa T, Endo N, Mori H. Racemization of Asp23 residue affects the aggregation properties of Alzheimer amyloid beta protein analogues. J Biol Chem. 1994; 269: 10205 -10208. [PubMed] .
- 61. Murakami K, Uno M, Masuda Y, Shimizu T, Shirasawa T, Irie K. Isomerization and/or racemization at Asp23 of Abeta42 do not increase its aggregative ability, neurotoxicity, and radical productivity in vitro. Biochem Biophys Res Commun. 2008; 366: 745 -751. [PubMed] .
- 62. Shimizu T, Watanabe A, Ogawara M, Mori H, Shirasawa T. Isoaspartate formation and neurodegeneration in Alzheimer's disease. Arch Biochem Biophys. 2000; 381: 225 -234. [PubMed] .
- 63. Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron. 1995; 14: 457 -466. [PubMed] .
- 64. Kuo YM, Emmerling MR, Woods AS, Cotter RJ, Roher AE. Isolation, chemical characterization, and quantitation of A beta 3-pyroglutamyl peptide from neuritic plaques and vascular amyloid deposits. Biochem Biophys Res Commun. 1997; 237: 188 -191. [PubMed] .
- 65. Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry. 2003; 42: 2768 -2773. [PubMed] .
- 66. Kumar S, Rezaei-Ghaleh N, Terwel D, Thal DR, Richard M, Hoch M, Mc Donald JM, Wullner U, Glebov K, Heneka MT, Walsh DM, Zweckstetter M, Walter J. Extracellular phosphorylation of the amyloid beta-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer's disease. EMBO J. 2011; 30: 2255 -2265. [PubMed] .
- 67. Milton NG. Phosphorylated amyloid-beta: the toxic intermediate in alzheimer's disease neurodegeneration. Subcell Biochem. 2005; 38: 381 -402. [PubMed] .
- 68. Milton NG. Phosphorylation of amyloid-beta at the serine 26 residue by human cdc2 kinase. Neuroreport. 2001; 12: 3839 -3844. [PubMed] .
- 69. Guntert A, Dobeli H, Bohrmann B. High sensitivity analysis of amyloid-beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience. 2006; 143: 461 -475. [PubMed] .
- 70. Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch Biochem Biophys. 1993; 301: 41 -52. [PubMed] .
- 71. Sergeant N, Bombois S, Ghestem A, Drobecq H, Kostanjevecki V, Missiaen C, Wattez A, David JP, Vanmechelen E, Sergheraert C, Delacourte A. Truncated beta-amyloid peptide species in pre-clinical Alzheimer's disease as new targets for the vaccination approach. J Neurochem. 2003; 85: 1581 -1591. [PubMed] .
- 72. Pike CJ, Overman MJ, Cotman CW. Amino-terminal deletions enhance aggregation of beta-amyloid peptides in vitro. J Biol Chem. 1995; 270: 23895 -23898. [PubMed] .
- 73. Millucci L, Ghezzi L, Bernardini G, Santucci A. Conformations and biological activities of amyloid beta peptide 25-35. Curr Protein Pept Sci. 2010; 11: 54 -67. [PubMed] .
- 74. Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter-Paier B, Prokesch M, Windisch M, Jagla W, Schlenzig D, Lindner C, Rudolph T, Reuter G, Cynis H, Montag D, Demuth HU, Rossner S. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer's disease-like pathology. Nat Med. 2008; 14: 1106 -1111. [PubMed] .
- 75. Schlenzig D, Manhart S, Cinar Y, Kleinschmidt M, Hause G, Willbold D, Funke SA, Schilling S, Demuth HU. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry. 2009; 48: 7072 -7078. [PubMed] .
- 76. Fabian H, Szendrei GI, Mantsch HH, Greenberg BD, Otvos L Jr. Synthetic post-translationally modified human A beta peptide exhibits a markedly increased tendency to form beta-pleated sheets in vitro. Eur J Biochem. 1994; 221: 959 -964. [PubMed] .
- 77. Saito T, Takaki Y, Iwata N, Trojanowski J, Saido TC. Alzheimer's disease, neuropeptides, neuropeptidase, and amyloid-beta peptide metabolism. Sci Aging Knowledge Environ. 2003; 2003: E1 .
- 78. Schilling S, Lauber T, Schaupp M, Manhart S, Scheel E, Bohm G, Demuth HU. On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry. 2006; 45: 12393 -12399. [PubMed] .
- 79. Kuo YM, Webster S, Emmerling MR, De LN, Roher AE. Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of A beta peptides of Alzheimer's disease. Biochim Biophys Acta. 1998; 1406: 291 -298. [PubMed] .
- 80. Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995; 80: 225 -236. [PubMed] .
- 81. Johnson LN and Barford D. The effects of phosphorylation on the structure and function of proteins. Annu Rev Biophys Biomol Struct. 1993; 22: 199 -232. [PubMed] .
- 82. Redegeld FA, Caldwell CC, Sitkovsky MV. Ecto-protein kinases: ecto-domain phosphorylation as a novel target for pharmacological manipulation? Trends Pharmacol Sci. 1999; 20: 453 -459. [PubMed] .
- 83. Kubler D and Barnekow A. Ecto-protein kinase activities in normal and transformed cells. Eur J Cell Biol. 1986; 40: 58 -63. [PubMed] .
- 84. Shaltiel S, Schvartz I, Korc-Grodzicki B, Kreizman T. Evidence for an extra-cellular function for protein kinase A. Mol Cell Biochem. 1993; 127-128: 283 -291. [PubMed] .
- 85. Walter J, Kinzel V, Kubler D. Evidence for CKI and CKII at the cell surface. Cell Mol Biol Res. 1994; 40: 473 -480. [PubMed] .
- 86. Kubler D, Pyerin W, Bill O, Hotz A, Sonka J, Kinzel V. Evidence for ecto-protein kinase activity that phosphorylates Kemptide in a cyclic AMP-dependent mode. J Biol Chem. 1989; 264: 14549 -14555. [PubMed] .
- 87. Rodriguez P, Mitton B, Kranias EG. Phosphorylation of glutathione-S-transferase by protein kinase C-alpha implications for affinity-tag purification. Biotechnol Lett. 2005; 27: 1869 -1873. [PubMed] .
- 88. Walter J, Schnolzer M, Pyerin W, Kinzel V, Kubler D. Induced release of cell surface protein kinase yields CK1- and CK2-like enzymes in tandem. J Biol Chem. 1996; 271: 111 -119. [PubMed] .
- 89. el-Moatassim C, Dornand J, Mani JC. Extracellular ATP and cell signalling. Biochim Biophys Acta. 1992; 1134: 31 -45. [PubMed] .
- 90. Dubyak GR and el-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am J Physiol. 1993; 265: C577 -C606. [PubMed] .
- 91. Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov. 2008; 7: 575 -590. [PubMed] .
- 92. Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview. Trends Neurosci. 2009; 32: 19 -29. [PubMed] .
- 93. Fujii S. ATP- and adenosine-mediated signaling in the central nervous system: the role of extracellular ATP in hippocampal long-term potentiation. J Pharmacol Sci. 2004; 94: 103 -106. [PubMed] .
- 94. Gourine AV, Dale N, Llaudet E, Poputnikov DM, Spyer KM, Gourine VN. Release of ATP in the central nervous system during systemic inflammation: real-time measurement in the hypothalamus of conscious rabbits. J Physiol. 2007; 585: 305 -316. [PubMed] .
- 95. Melani A, Turchi D, Vannucchi MG, Cipriani S, Gianfriddo M, Pedata F. ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem Int. 2005; 47: 442 -448. [PubMed] .
- 96. Iijima K, Ando K, Takeda S, Satoh Y, Seki T, Itohara S, Greengard P, Kirino Y, Nairn AC, Suzuki T. Neuron-specific phosphorylation of Alzheimer's beta-amyloid precursor protein by cyclin-dependent kinase 5. J Neurochem. 2000; 75: 1085 -1091. [PubMed] .
- 97. Suzuki T and Nakaya T. Regulation of amyloid beta-protein precursor by phosphorylation and protein interactions. J Biol Chem. 2008; 283: 29633 -29637. [PubMed] .
- 98. Walter J, Schindzielorz A, Hartung B, Haass C. Phosphorylation of the beta-amyloid precursor protein at the cell surface by ectocasein kinases 1 and 2. J Biol Chem. 2000; 275: 23523 -23529. [PubMed] .
- 99. Walter J, Fluhrer R, Hartung B, Willem M, Kaether C, Capell A, Lammich S, Multhaup G, Haass C. Phosphorylation regulates intracellular trafficking of beta-secretase. J Biol Chem. 2001; 276: 14634 -14641. [PubMed] .
- 100. von Arnim CA, Tangredi MM, Peltan ID, Lee BM, Irizarry MC, Kinoshita A, Hyman BT. Demonstration of BACE (beta-secretase) phosphorylation and its interaction with GGA1 in cells by fluorescence-lifetime imaging microscopy. J Cell Sci. 2004; 117: 5437 -5445. [PubMed] .
- 101. Walter J, Capell A, Grunberg J, Pesold B, Schindzielorz A, Prior R, Podlisny MB, Fraser P, Hyslop PS, Selkoe DJ, Haass C. The Alzheimer's disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol Med. 1996; 2: 673 -691. [PubMed] .
- 102. De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P, George-Hyslop PS, Van LF. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer's disease-associated presenilins. J Biol Chem. 1997; 272: 3590 -3598. [PubMed] .
- 103. Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986; 83: 4913 -4917. [PubMed] .
- 104. Delobel P, Flament S, Hamdane M, Mailliot C, Sambo AV, Begard S, Sergeant N, Delacourte A, Vilain JP, Buee L. Abnormal Tau phosphorylation of the Alzheimer-type also occurs during mitosis. J Neurochem. 2002; 83: 412 -420. [PubMed] .
- 105. Mi K and Johnson GV. The role of tau phosphorylation in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2006; 3: 449 -463. [PubMed] .
- 106. Chun W and Johnson GV. The role of tau phosphorylation and cleavage in neuronal cell death. Front Biosci. 2007; 12: 733 -756. [PubMed] .
- 107. Moloney A, Sattelle DB, Lomas DA, Crowther DC. Alzheimer's disease: insights from Drosophila melanogaster models. Trends Biochem Sci. 2010; 35: 228 -235. [PubMed] .
- 108. Crowther DC, Page R, Rival T, Chandraratna DS, Lomas DA. Using a Drosophila model of Alzheimer's disease. SEB Exp Biol Ser. 2008; 60: 57 -77. [PubMed] .
- 109. Walker LC, Bian F, Callahan MJ, Lipinski WJ, Durham RA, LeVine H. Modeling Alzheimer's disease and other proteopathies in vivo: is seeding the key? Amino Acids. 2002; 23: 87 -93. [PubMed] .
- 110. Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006; 313: 1781 -1784. [PubMed] .
- 111. LeVine H III. The Amyloid Hypothesis and the clearance and degradation of Alzheimer's beta-peptide. J Alzheimers Dis. 2004; 6: 303 -314. [PubMed] .
- 112. Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, Fernandez CO, Schmid A, Chegini F, Gai WP, Chiappe D, Moniatte M, Schneider BL, Aebischer P, Eliezer D, Zweckstetter M, Masliah E, Lashuel HA. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci. 2010; 30: 3184 -3198. [PubMed] .
- 113. Liang FC, Chen RP, Lin CC, Huang KT, Chan SI. Tuning the conformation properties of a peptide by glycosylation and phosphorylation. Biochem Biophys Res Commun. 2006; 342: 482 -488. [PubMed] .
- 114. Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, Olschewski D, Yin G, Zweckstetter M, Masliah E, Kahle PJ, Hirling H, Lashuel HA. Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem. 2010; 285: 2807 -2822. [PubMed] .
- 115. Cavallarin N, Vicario M, Negro A. The role of phosphorylation in synucleinopathies: focus on Parkinson's disease. CNS Neurol Disord Drug Targets. 2010; 9: 471 -481. [PubMed] .