



Introduction
Regulation of calcium homeostasis and bone metabolism is the most widely recognized function of 1,25-dihydroxyvitamin D3 also known as calcitriol (1,25-VD), the hormonal form of vitamin D. However, there is a growing body of evidence showing that 1,25-VD also plays a significant role in the prevention of different types of cancer [1-9]. The molecular mechanisms that may be involved in the chemo-preventive and antitumor activities of 1,25-VD have been extensively reviewed in recent publications [3,9,10]. One of the mechanisms that may contribute to the chemo-preventive properties of 1,25-VD is modulation of the DNA damage response that facilitates successful DNA repair after treatment with genotoxic agents [10-16]. This mechanism was observed particularly after exposure of cells to UV light [11-14]. Recent studies on gene profiling revealed that the DNA repair genes are among a multitude of genes whose transcription is induced by 1,25-VD [17-19].
Another mechanism that may contribute to the chemo-preventive properties of 1,25-VD is likely provided by its antioxidant activity. Persistent DNA damage by endogenous oxidants, by-products of aerobic respiration, is considered to be one of the causes of carcinogenesis and aging [20-24]. Accumulated genomic mutations from improperly repaired DNA, particularly at the sites of oncogenes or tumor suppressor genes, may result in malignant transformation. There is ample evidence that 1,25-VD provides protection against oxidative DNA damage. Thus, constitutive DNA damage in colon epithelium, likely induced by endogenous oxidants, was seen to be elevated in vitamin D receptor (VDR)-knockout mice [25]. Conversely, the treatment of humans with a daily dose of 800 I.U. of vitamin D was shown to reduce oxidative DNA damage in colon epithelium [26]. Treatment of cells of several cell lines with 1,25-VD led to induction of enzymes involved in protection against oxidative damage. One of them is thioredoxin reductase 1 (TXNRD1), the enzyme which maintains thioredoxin in the reduced state needed to confer a reduced intracellular redox state and thereby limit intracellular oxidant stress [27-30]. Another antioxidant protein induced by 1,25-VD is superoxide dismutase (SOD) [28-31].
We have recently reported that untreated normal or tumor cells in culture exhibit a “background” level of DNA damage signaling (DDS) revealed as histone H2AX phosphorylation on Ser139 and activation of Ataxia Telangiectasiamutated protein kinase (ATM) through phosphorylation on Ser1981 [32-34]. These phosphorylation events, which report DNA damage and in particular formation of DNA double-strand breaks (DSBs) [35,36], were detected in individual cells immunocytochemically using phospho-specific Abs and measured by flow and laser scanning cytometry (LSC) [32-34]. Multiparameter cytometric analysis made it possible to correlate the level of this intrinsic, constitutive DDS, with the cell cycle phase and with the cell's propensity to undergo apoptosis. In a series of experiments we demonstrated that, at least to a large degree, the constitutive DDS is in response to oxidative DNA damage induced by endogenous oxidants produced during aerobic respiration [32-34]. Mitogenic stimulation of lymphocytes dramatically increased the level of constitutive DDS concurrent with an increase in the generation of endogenous oxidants and activation of the mTOR pathway [37]. Conversely, prolonged exposure of cells to agents that reduced the level of endogenous oxidants such as hyaluronan [38], or to metformin, the anti-diabetic drug, inducer of AMP-activated protein kinase and mTOR suppressor [39], led to a significant attenuation of constitutive DDS. In light of previous evidence for the antioxidant and chemo-preventive properties of 1,25-VD, in the present study we explored whether this vitamin/hormone has the ability to affect the level of constitutive DDS.
Results
Exposure of A549 cells to 1,25-VD at a concentration of 2 or 10 nM for 24 and 48 h led to a distinct reduction in the level of expression of γH2AX (Figure 1). As it is evident from the bivariate DNA content versus γH2AX expression dotplots, the degree of decline in γH2AX expression was similar regardless of 1,25-VD concentration and duration of cell treatment. However, a distinct difference was apparent with regard to cell cycle phase. The cells most affected were in S-phase of the cell cycle which showed a reduction in γH2AX within the range of 36% to 39%. The effect of 1,25D was less pronounced in the case of G1 or G2M phase cells as their γH2AX expression was decreased by only 18-22% or 11-21%, respectively. During the time and at the concentration of 1,25-VD studied here, there was no detectable effect on the cell cycle distribution of A549 cells, as is evident from the similarity of the cellular DNA content histograms shown in the respective insets.
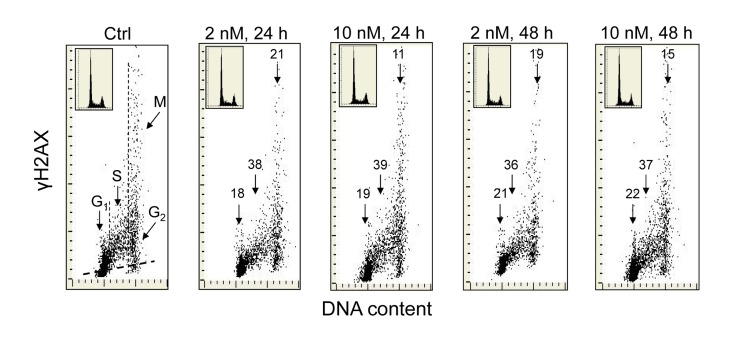
Figure 1. Effect of the treatment of A549 cells with 1,25-VD on the level of expression of γH2AX with respect to the cell cycle phase Exponentially growing human pulmonary carcinoma A549 cells were untreated (Ctrl) or treated with 2 or 10 nM 1,25-VD for 24 or 48 h and expression of γH2AX was detected immunocytochemically using phospho-specific Ab targeting the phosphorylated S139 epitope; the intensity of fluorescence was measured by laser scanning cytometry (LSC) [70]. The numbers above the respective arrows indicate the percent reduction in intensity of immunofluorescence of the mean values of cell populations in G1, S and G2M phases of the cell cycle (gated as shown in the Ctrl panel) in the 1,25-VD-treated cells with respect to the untreated cells (Ctrl), in the same phase of cell cycle. Inserts show cellular DNA content histograms from the respective cultures. The skewed dash line in Ctrl panel shows the maximal level of fluorescence intensity of the cells stained with isotype control Ab.
Figure 2 illustrates the effect of 1,25-VD on the level of phosphorylation of ATM at S1981 in A549 cells. As in the case of H2AX phosphorylation (Figure 1), the expression of S1981 phosphorylated ATM was markedly reduced in cells exposed to 1,25-VD. The degree of decline in expression of phosphorylated ATM was similar at 2 nM and at 10 nM of vitamin D and following 24 h or 48 h of exposure. As compared with the effect of 1,25-VD on γH2AX expression (Figure 1) there were less pronounced differences in the degree of reduction of ATM-S1981P between the cells in different phases of the cell cycle. However, the cells in S phase were somewhat more affected (33% - 43% reduction) compared to G1 (30-35%) or G2M cells (17-30%), respectively.
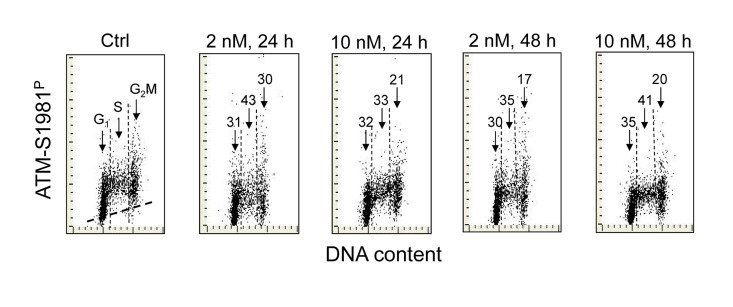
Figure 2. Effect of treatment of A549 cells with 1,25-VD on the level of expression of ATM phosphorylated on Ser1981 in relation to the cell cycle phase Expression of ATM-S1981P in A549 cells treated with 2 nM or 10 nM 1,25-VD for 24 or 48 h was detected immunocytochemically using phospho-specific Abs and measured by LSC. The numbers above the arrows indicate the percent reduction in intensity of immunofluorescence of the mean values of cell populations in G1, S and G2M phases of the cell cycle in the 1,25-VD-treated cells with respect to the untreated cells (Ctrl) in the same phase of cell cycle. The skewed dash line in Ctrl panel shows the maximal level of fluorescence intensity of the cells stained with isotype control Ab.
Treatment of human B lymphoblastoid TK6 cells with 1,25-VD also led to a decrease in the level of constitutive expression of γH2AX and ATM-S1981P (Figure 3). The overall effect of 1,25-VD in suppression of the level of constitutive phosphorylation of these proteins in TK6 cells was somewhat weaker than in A549 cells (Figures 1 and 2). However, as in the case of A549 cells, the S-phase cells were more affected by 1,25-VD than G1 or G2M phase cells in TK6 cultures and the suppressive effect of 1,25-VD on H2AX phosphorylation (13-18%) was more pronounced than on phosphorylation of ATM (8-12%).
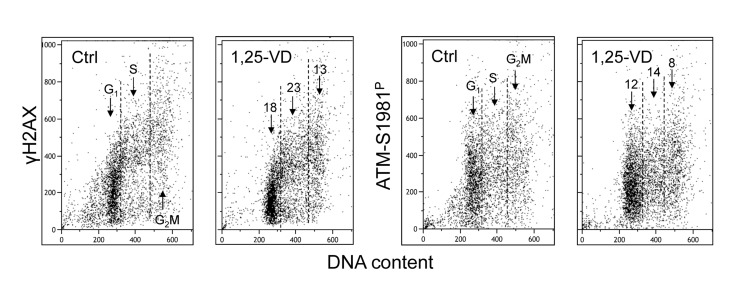
Figure 3. Effect of treatment of TK6 cells with 1,25-VD on the level of constitutive expression of γH2AX and ATM-S1981P Exponentially growing TK6 cells were untreated (Ctrl) or treated with 10 nM 1,25-VD for 24 h. Expression of γH2AX and ATM-S1981P was detected in individual cells immunocytochemically, cellular fluorescence was measured by flow cytometry. The numbers above the arrows show the percent reduction in intensity of immunofluorescence of the mean values of cell populations in G1, S and G2M phases of the cell cycle in the 1,25-VD-treated cells with respect to the untreated cells (Ctrl) in the same phase of cell cycle.
We have also tested the effect of 1,25-VD on the level of constitutive expression of γH2AX and ATM-S1091P in proliferating human lymphocytes (Figure 4). In the initial experiments we noticed that when 1,25-VD was added into cultures concurrent with PHA, lymphocyte stimulation, as expressed by the increases in cellular RNA content and progression through the cell cycle, was delayed compared to cells growing in the absence of 1,25-VD. This observation was consistent with prior studies that showed the suppressive effect of 1,25-VD on lymphocyte stimulation by a variety of mitogens [41-43]. However, when lymphocytes were already fully in the cell cycle 48 h following stimulation by PHA, the treatment with 1,25-VD did not alter their cell cycle progression over the subsequent 24 h. Under such conditions, we were able to test the effect of 1,25-VD on constitutive expression of γH2AX and ATM-S1981P on cells which had fully entered the cell cycle similar to the cells stimulated by PHA growing in the absence of 1,25-VD (Figure 4A). Therefore, the results were not biased by the anti-proliferative effect of 1,25D which would have been apparent if 1,25-VD had been added earlier during lymphocytes stimulation by PHA.
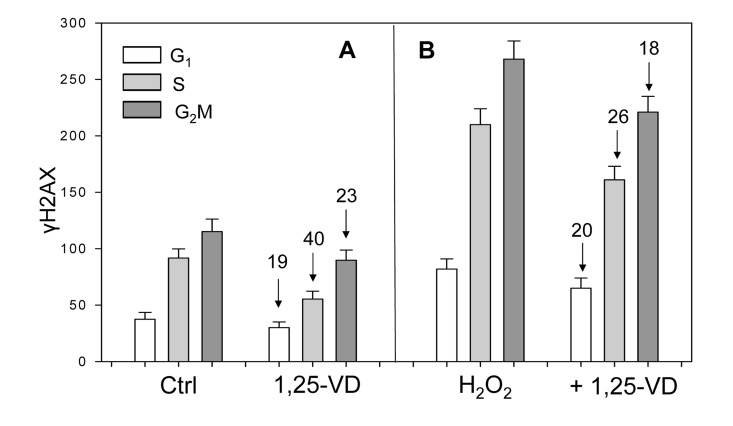
Figure 4. Effect of treatment of mitogenically stimulated proliferating human lymphocytes untreated or treated with H2O2 and 1,25-VD on the level of expression of γH2AX Human peripheral blood lymphocytes mitogenically stimulated with PHA for 48 h were untreated (Ctrl) or treated with 10 nM 1,25-VD for 24 h (A). Cells in another set of cultures (B) were either untreated with 1,25-VD but exposed to 200 μM of H2O2 for 1 h (H2O2) or were pretreated with 1,25-VD for 24 h and then treated with 200 μM H2O2 for 1 h (+H2O2). Expression of γH2AX was detected immunocytochemically and cell fluorescence was measured by flow cytometry. Through gating analysis (as shown in Figs. 1-3) the mean values (+SD) of fluorescence intensity for cells in G1, S and G2M phases of the cell cycle were obtained and plotted as bar graphs. Numbers above the arrows show the percent decline of the mean values of the 1,25-VD-treated cells compared to the cells not treated with 1,25-VD in the respective phases of the cycle.
Unlike the results on A549 and TK6 cells (Figures 1-3, scatterplots), the data on lymphocytes are presented as bar graphs (Figure 4). The effect of 1,25-VD on lymphocytes was similar to that its effect on A549 or TK6 cells. As it is evident, the level of expression of γH2AX was distinctly lower in the 1,25-VD treated cells (Fig 4A). The cells most affected were in S-phase, where γH2AX fluorescence was decreased by 40%, nearly twice as much as that of G1 or G2M cells. The level of expression of ATM-S1981P was also decreased in 1,25-VD treated lymphocytes, but the decrease was less pronounced (10-18% across all phases of the cell cycle) compared to that of γH2AX (data not shown).
In another set of lymphocyte cultures, we explored whether 1,25-VD can affect expression of γH2AX upon induction of oxidative damage by H2O2. Towards this end, lymphocytes stimulated by PHA for 48 h and then treated with 1,25-VD for an additional 24 h were exposed to 200 μM H2O2 (+ 1,25-VD) for 60 min; parallel cultures of PHA stimulated lymphocytes were exposed to 200 μM H2O2 for 60 min without pretreatment with 1,25-VD (H2O2) (Figure 4B). The cells treated only with H2O2 had approximately a two-fold higher level of γH2AX expression compared with the untreated cells (Ctrl), consistent with the induction of oxidative DNA damage by the peroxide [40]. However, the effect of treatment with H2O2 in terms of a decline in expression of γH2AX was less pronounced in the cells exposed to 1,25-VD. Specifically, compared with the cells not treated with 1,25-VD, the treated cells exhibited an 18-26% lower level of γH2AX expression in which the S phase cells were more affected compared to G1 or G2M phase cells.
In addition to measuring the effect of 1,25-VD on expression of γH2AX and ATM-S1981P in lymphocytes we have also measured the effect of this hormone on the intracellular level of ROS as detected by the lymphocytes' ability to oxidize dihydrofluorescein. This reagent (2,7-dichlorodihydrofluorescein diacetate; DCFH-DA) is permeant and non-fluorescent but when oxidized within the cell becomes fluorescent. Exposure of PHA-stimulated lymphocytes to 10 nM 1,25-VD for 24 h and 48 h (as described in Materials and Methods) reduced by about half (from 62 ± 3.5 to 32 ± 4.7 AU) or by nearly three-fold (from 62 ± 3.5 to 23 ± 4.7) their ability to oxidize DCFH-DA, respectively. However, we were not able to observe a significant reduction of DCFH-DA fluorescence in TK6 or A549 cells treated for 24 h with 2 nM or 10 nM 1,25-VD (data not shown).
Discussion
The present data show that the level of constitutive DNA damage signaling as measured by expression of γH2AX and ATM-S1981P was decreased in A549 and TK6 tumor cells cultured in the presence of 1,25-VD for 24 - 48 h. Also decreased was the level of γH2AX and ATM-S1981P in proliferating normal human lymphocytes cultured in the presence of 1,25-VD. These data suggest that both in normal proliferating lymphocytes as well as in A549 or TK6 tumor cells the level of constitutive DNA damage, likely induced by-products of oxidative metabolism was attenuated by 1,25-VD. Several mechanisms may be responsible for this effect.
It has been observed that the level of constitutive DNA damage signaling is more pronounced in S and G2M than in G1-phase cells [32-34]. This level was also seen to dramatically increase during mitogenic stimulation of lymphocytes [37]. Most likely, the high intensity of constitutive DNA damage signaling in S-phase cells is reporting DNA replication stress that occurs when replication forks encounter sites of DNA with oxidative damage. Replication stress, especially combined with mTOR activation can lead to elevated levels of DNA damage signaling (“pseudo-DNA damage response”) associated with cell senescence [39,44-47]. The suppressive effect on H2AX and ATM phosphorylation by 1,25-VD, as is evident in S-phase cells, suggests that in cells exposed to this hormone the intensity of replication stress is greatly diminished. Consistent with this is the evidence that in some cell types 1,25-VD suppresses Akt/mTOR signaling [48-50]. It should be noted, however, that in leukemic cells triggered by 1,25-VD to differentiation, the PI3K/Akt pathway is actually activated, making the association between these pathways and biological effects complex [51,52]. It is possible that activation of the PI3K/AKT/mTOR pathway takes place exclusively after induction of differentiation of promyelocytic leukemic (HL-60) cells whereas in other cell systems, when differentiation is not apparent, mTOR is inhibited by this hormone. Interestingly, inhibition of mTORC1 by the rapamycin analog RAD001 was shown to potentiate the effect of 1,25-VD to induce growth arrest and differentiation in AML cells, which prompted the authors to propose the strategy of concomitant administration of both 1,25-VD and an mTOR inhibitor in treatment of AML [53]. The anti-aging properties of vitamin D [54-58] would be consistent with its mTOR inhibitory activity [47,59].
The attenuation of DNA damage signaling by 1,25-VD in lymphocytes as seen in the present study would also be consistent with the anti-oxidant properties of this hormone [25-31]. Indeed we observed that exposure of lymphocytes to 1,25-VD led to a decrease in abundance of reactive oxidants that were detected by their ability to oxidize DCFH-DA. We were unable, however, to see a significant effect following exposure of TK6 or A549 cells for 24 h to 1,25-VD on oxidation of DCFH-DA. Yet, at the same time a reduction in the constitutive level of γH2AX in these cells was observed. While it is possible that the sensitivity of the DCFH-DA-ROS detection assay is inadequate to reveal minor changes in abundance of endogenous oxidants that still affect the level of constitutive H2AX phosphorylation, factors other than ROS induced by 1,25-VD may contribute to the lowering DNA damage signaling.
Our present findings are in accord with the recently published data of Ting et al., [60] in a mouse model system which show that 1,25-VD reduces the level of H2AX phosphorylation and ATM activation after induction of DNA damage by H2O2 or by N-nitroso-N-methylurea (NMU). Of interest are also their findings that 1,25-VD enhanced expression of DNA repair genes ATM and RAD50 and protected cells from genotoxic stress and malignant transformation. The mechanism advanced by the authors involves the cross-talk between ATM and 1,25-VD receptor (VDR). Specifically, activation of ATM, the early step in the DNA damage response pathway, in the presence of 1,25-VD leads to phosphorylation of VDR, which in turn induces transcriptional upregulation of ATM and RAD50 thereby promoting effective DSB repair [60]. With such interactions between ATM and VDR, one would expect that 1,25-VD may affect downstream pathways such as those leading to apoptosis and cell senescence, thereby providing a barrier to cancer development, and protecting genome integrity. The interactions between ATM and VDR may also be the mechanism that explains our data on reduction of constitutive H2AX phosphorylation (likely reporting the presence of DSBs) in A549 and TK6 cells in the absence of detectable effect of 1,25-VD on ROS. According to this mechanism [60] it is feasible that constitutive activation of ATM in the presence of 1,25-VD induces phosphorylation of VDR which further enhances expression of ATM and RAD50, leads to a more effective DNA repair, reduces frequency of DSBs and, as a consequence, dampens down the level of DNA damage signaling (γH2AX expression).
It has been reported that proliferation of cells from various solid tumor types including malignant melanoma [61], colon [62], breast [64] and prostate cancer [64] is inhibited by 1,25-VD. Vitamin D and its analogues are also widely recognized as potential anticancer drugs in the treatment of some hematological malignancies [2,9]. It is unclear if there is an association between molecular mechanisms responsible for the presently observed attenuation of constitutive DNA damage signaling and the previously reported effects of 1,25-VD on cell survival. For instance, in myeloid leukemia cells in culture 1,25-VD affects cell survival by upregulation of the AKT [52], and the KSR2 pathways [65]. Most recently, the upregulation of microRNA32 expression by 1,25-VD was shown to control the levels of the pro-apoptotic protein BIM [66]. These molecules could be components of a complex network that orchestrates cell survival at the level of DNA damage. However, the attenuation of DNA damage response (DDR) by 1,25-VD demonstrated here illustrates that DDR can be a barrier, or a pathway, to tumor progression, depending on the presence and nature of extracellular factors [67,68].
Materials and Methods
Cells, Cell Treatment
Human lung carcinoma A549 cells, were obtained from the American Type Culture Collection (ATCC, Manassas,VA). Human B-cell lymphoblastoid TK6 cells were kindly provided by Dr Howard Liber of Colorado State University (Fort Collins CO). Human peripheral blood lymphocytes were obtained by venipuncture from healthy volunteers following informed consent according to Institutional guidelines, and isolated by density gradient centrifugation. A549 cells were cultured in Ham's F12K, TK6 and lymphocytes were cultured in RPMI 1640 with 2 mM L-glutamine adjusted to contain 1.5 g/L sodium bicarbonate supplemented with 10% fetal bovine serum (GIBCO/Invitrogen, Carlsbad, CA). Adherent A549 cells were grown in dual-chambered slides (Nunc Lab-Tek II), seeded with 105 cells/ml suspended in 2 ml medium per chamber. TK6 cells and lymphocytes were grown in suspension; lymphocyte cultures were treated with the polyvalent mitogen phytohemaglutinin (Sigma/Aldrich; St Louis, MO) as described [37]. The active form of vitamin D, 1,25-VD, was kindly provided by Dr. Milan Uskokovic, and the stock solution was maintained in ethanol. During treatment with 1,25-VD the cells were in exponential phase of growth; control cells were treated with an equivalent concentration of ethanol. After exposure to 1,25-VD at various concentrations and for specified periods of time (as shown in figure legends) the cells were rinsed with phosphate buffered salt solution (PBS) and fixed in 1% methanol-free formaldehyde (Polysciences, Warrington, PA) for 15 min on ice The cells were then transferred to 70% ethanol and stored at -20 °C for up to 3 days until staining.
Detection of H2AX Phosphorylation and ATM Activation
The cells were washed twice in PBS and with 0.1% Triton X-100 (Sigma) in PBS for 15 min and with a 1% (w/v) solution of bovine serum albumin (BSA; Sigma) in PBS for 30 min to suppress nonspecific antibody (Ab) binding. The cells were then incubated in 1% BSA containing a 1:300 dilution of phospho-specific (Ser139) γH2AX mAb (Biolegend, San Diego, CA) or with a 1:100 dilution of phospho-specific (Ser1981) ATM mAb (Millipore, Tamecula, CA). The secondary Ab was tagged with AlexaFluor 488 fluorochrome (Invitrogen/Molecular Probes, used at 1:200 dilution). Cellular DNA was counterstained with 2.8 μg/ml 4,6-diamidino-2-phenylindole (DAPI; Sigma). Each experiment was performed with an IgG control in which cells were labeled only with the secondary AlexaFluor 488 Ab, without primary Ab incubation to estimate the extent of nonspecific adherence of the secondary Ab to the cells. The fixation, rinsing and labeling of A549 cells was carried out on slides, and lymphocytes and TK6 cells in suspension. Other details have been previously described [39,40].
Reactive Oxygen Species (ROS) Detection
Untreated cells as well as cells treated with vitamin D were incubated 60 min with 10 μM 2',7'-dihydro-dichlorofluorescein-diacetate (H2DCF-DA) (Invitrogen/ Molecular Probes) at 37°C. Cellular green fluorescence was then measured by flow cytometry. Following oxidation by ROS and peroxides within cells the non-fluorescent substrate H2DCF-DA is converted to the highly fluorescent derivative DCF [59].
Analysis of Cellular Fluorescence
A549 cells: Cellular immunofluorescence representing the binding of the respective phospho-specific Abs as well as the blue emission of DAPI stained DNA was measured by Laser Scanning Cytometry (LSC) [70] (iCys; CompuCyte, Westwood, MA) utilizing standard filter settings; fluorescence was excited with 488-nm argon, helium-neon (633 nm) and violet (405 nm) lasers. The intensities of maximal pixel and integrated fluorescence were measured and recorded for each cell. At least 3,000 cells were measured per sample. Gating analysis was carried out as described in Figure legends. TK6 cells and lymphocytes: Cellular fluorescence was measured by using a MoFlo XDP (Beckman-Coulter, Brea, CA) high speed flow cytometer/sorter. DAPI fluorescence was excited with the UV laser (355-nm), AlexaFluor 488 and DCF with the argon ion (488-nm) laser. All experiments were repeated at least three times
Acknowledgments
Supported by NCI RO1 28 704 and by the Andrew Welke Foundation for Cancer Research.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Garland CF, Garland FC, Gorham ED, Lipkin M, Newmark H, Mohr SB, Holick MF. The role of vitamin D in cancer prevention. Am J Publ Health. 2006; 96: 252 -261. .
- 2. Gocek E and Studzinski GP. Vitamin D and differentiation in cancer. Crit Rev Clin Lab Sci. 2009; 46: 190 -209. [PubMed] .
- 3. Krishnan AV, Trump DL, Johnson CS, Feldman D. The role of vitamin D in cancer prevention and treatment. Endocrinol Metab Clin N Am. 2010; 39: 401 -418. .
- 4. Fleet JC, Desmet M, Johnson R, Li Y. Vitamin D and cancer: a review of molecular mechanisms. Biochem J. 2012; 221: 61 -76. [PubMed] .
- 5. Zinser GM, Suckow M, Welsh J. Vitamin D receptor (VDR) ablation alters carcinogen-induced tumorigenesis in mammary gland, epidermis and lymphoid tissues. J Steroid Biochem Mol Biol. 2005; 97: 153 -164. [PubMed] .
- 6. Trump DL, Deeb KK, Johnson CS. Vitamin D: considerations in the continued development as an agent for cancer prevention and therapy. Cancer J. 2010; 16: 1 -9. [PubMed] .
- 7. Giardina C, Madigan JP, Godman Tierney CA, Brenner BM, Rosenberg DW. Vitamin D resistance and colon cancer prevention. Carcinogenesis. (Epub) .
- 8. Weisman Y. Non-classic unexpected functions of vitamin D. Pediatr Endocrin Rev. 2010; 8: 103 -107. .
- 9. Studzinski GP, Wang X, Ji Y, Wang Q, Zhang Y, Kutner A, Harrison JS. The rationale for deltanoids in therapy for myeloid leukemia: role of KSR-MAPK-C/EBP pathway. J Steroid Biochem Mol Biol. 2005; 97: 47 -55. [PubMed] .
- 10. Ting HJ, Yasmin-Karim S, Yan S-J, Hsu JW, Lin T-H, Zeng W, Messing J, Sheu T-J, Bao B-Y, Li W, Messing E, Lee Y-F. A positive feedback signaling loop between ATM and the vitamin D receptor is critical for cancer chemoprevention by vitamin D. Cancer Res. 2012; 72: 958 -968. [PubMed] .
- 11. De Haes P, Garmyn M, Verstuyf A, De Clercq P, Vandewalle M, Degreef H, et al. 1,25-Dihydroxyvitamin D3 and analogues protect primary human keratinocytes against UVB-induced DNA damage. J Photochem Photobiol B. 2005; 78: 141 -148. [PubMed] .
- 12. Wong G, Gupta R, Dixon KM, Deo SS, Choong SM, Halliday GM, et al. 1,25-Dihydroxyvitamin D and three low-calcemic analogs decrease UV-induced DNA damage via the rapid response pathway. J Steroid Biochem Mol Biol. 2004; 89-90: 567 -570. [PubMed] .
- 13. Chatterjee M. Vitamin D and genomic stability. Mutat Res. 2001; 475: 69 -87. [PubMed] .
- 14. Hanada K, Sawamura D, Nakano H, Hashimoto I. Possible role of 1,25-dihydroxyvitamin D3-induced metallothionein in photoprotection against UVB injury in mouse skin and cultured rat keratinocytes. J Dermatol Sci. 1995; 9: 203 -208. [PubMed] .
- 15. De Haes P, Garmyn M, Degreef H, Vantieghem K, Bouillon R, Segaert S. 1,25-Dihydroxyvitamin D3 inhibits ultraviolet B-induced apoptosis, Jun kinase activation, and interleukin-6 production in primary human keratinocytes. J Cell Biochem. 2003; 89: 663 -673. [PubMed] .
- 16. Banakar MC, Paramasivan SK, Chattopadhyay MB, Datta S, Chakraborty P, Chatterjee M, et al. 1alpha, 25-dihydroxyvitamin D3 prevents DNA damage and restores antioxidant enzymes in rat hepatocarcinogenesis induced by diethylnitrosamine and promot-ed by phenobarbital. World J Gastroenterol. 2004; 0: 1268 -1275. [PubMed] .
- 17. Krishnan AV, Shinghal R, Raghavachari N, Brooks JD, Peehl DM, Feldman D. Analysis of vitamin D-regulated gene expression in LNCaP human prostate cancer cells using cDNA microarrays. Prostate. 2004; 59: 243 -251. [PubMed] .
- 18. Wu-Wong JR, Nakane M, Ma J, Ruan X, Kroeger PE. Effects of Vitamin D analogs on gene expression profiling in human coronary artery smooth muscle cells. Atherosclerosis. 2006; 186: 20 -28. [PubMed] .
- 19. Haussler MR, Haussler CA, Whitfield GK, Hsieh JC, Thompson PD, Barthel TK, Bartik L, Egan JB, Wu Y, Kubicek JL, Lowmiller CL, Moffet EW, Forster RE, Jurutka PW. The nuclear vitamin D receptor controls the expression of genes encoding factors which feed the “Fountain of Youth” to mediate healthful aging. J. Steroid Biochem. Mol Biol. 2010; 121: 88 -97. .
- 20. Barzilai A and Yamamoto K. DNA damage responses to oxidative stress. DNA repair (Amst). 2004; 3: 1109 -1115. [PubMed] .
- 21. Vilenchik MM and Knudson AG. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci USA. 2003; 100: 12871 -12876. [PubMed] .
- 22. Floyd RA, West M, Henssley K. Oxidative biochemical markers; clues to understanding aging in long-lived species. Exp Gerontol. 2001; 36: 619 -640. [PubMed] .
- 23. Marnett LJ, Riggins JN, West JD. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J Clin Invest. 2003; 111: 583 -593. [PubMed] .
- 24. Valko M, Rhodes C-J, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006; 160: 1 -40. [PubMed] .
- 25. Kallay E, Pietschmann P, Toyokuni S, Bajna E, Hahn P, Mazzucco K, Bieglmayer C, Kato S, Cross HS. Characterization of a vitamin D receptor knockout mouse as a model of colorectal hyperproliferation and DNA damage. Carcinogenesis. 2001; 22: 1429 -1435. [PubMed] .
- 26. Fedirko V, Bostick RM, Long Q, Flanders WD, McCullough ML, Sidelnikov E, Daniel CR, Rutherford RE, Shaukat A. Effects of supplemental vitamin D and calcium on oxidative DNA damage marker in normal colorectal mucosa: a randomized clinical trial. Cancer Epidemiol. Biomarkers Prev. 2010; 19: 280 -291. .
- 27. Schutze N, Fritzche J, Ebert-Dumig R, Schneider D, Kohrle J, Andreesen R, Kreutz M, Jakob F. The selenoprotein thioredoxin reductase is expressed in peripheral blood monocytes and THP1 human myeloid leukemia cells - regulation by 1,25-dihydroxyvitamin D3 and selenite. Biofactors. 1999; 10: 329 -338. [PubMed] .
- 28. Peehl DM, Shinghal R, Nonn L, Seto E, Krishnan AV, Brooks JD, Feldman D. Molecular activity of 1,25-dihydroxyvitamin D3 in primary cultures of human prostatic epithelial cells revealed by cDNA microarray analysis. J. Steroid Biochem Mol. Biol. 2004; 92: 131 -141. [PubMed] .
- 29. Swami S, Raghavachari N, Muller UR, Bao YP, Feldman D. Vitamin D growth inhibition of breast cancer cells: gene expression patterns assessed by cDNA microarray. Breast Cancer Res Treat. 2003; 80: 49 -62. [PubMed] .
- 30. Palmer HG, Sanchez-Carbayo M, Ordonez-Moran P, Larriba MJ, Cordon-Cardo C, Munoz A. Genetic signatures of differentiation induced by 1α,25-dihydroxyvitamin D3 in human colon cancer cells. Cancer Res. 2003; 63: 7799 -7806. [PubMed] .
- 31. Lambert JR, Kelly JA, Shim M, Huffer WE, Nordeen SK, Baek SJ, Eling TE, Lucia MS. Prostate derived factor in human prostate cancer cells: gene induction by vitamin D via a p53-dependent mechanism and inhibition of prostate cancer cell growth. J Cell Physiol. 2006; 208: 566 -574. [PubMed] .
- 32. Huang X, Tanaka T, Kurose A, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell-cycle phase specific and attenuated by scavenging reactive oxygen species. Int J Oncol. 2006; 29: 495 -501. [PubMed] .
- 33. Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006; 5: 1940 -1945. [PubMed] .
- 34. Zhao H, Tanaka T, Halicka HD, Traganos F, Zarebski M, Dobrucki J, Darzynkiewicz Z. Cytometric assessment of DNA damage by exogenous and endogenous oxidants reports the aging-related processes. Cytometry A. 2007; 71A: 905 -914. [PubMed] .
- 35. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998; 273: 5858 -5868. [PubMed] .
- 36. Bartkova J, Bakkenist CJ, Rajpert-De Meyts E, Skakkebaek NE, Sehested M, Lukas J, Kastan MB, Bartek J. ATM activation in normal human tissues and testicular cancer. Cell Cycle. 2005; 4: 838 -845. [PubMed] .
- 37. Tanaka T, Kajstura M, Halicka HD, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation are strongly amplified during mitogenic stimulation of lymphocytes. Cell Prolif. 2007; 40: 1 -13. [PubMed] .
- 38. Zhao H, Tanaka T, Mitlitski V, Heeter J, Balazs EA, Darzynkiewicz Z. Protective effect of hyaluronate on oxidative DNA damage in WI-38 and A549 cells. Int J Oncol. 2008; 32: 1159 -1169. [PubMed] .
- 39. Halicka HD, Zhao H, Li J, Traganos F, Zhang S, Lee M, Darzynkiewicz Z. Genome protective effect of metformin as revealed by reduced level of constitutive DNA damage signaling. Aging (Albany). 2011; 3: 1028 -1038. .
- 40. Zhao H, Dobrucki J, Rybak P, Traganos F, Halicka H.D, Darzynkiewicz Z. Induction of DNA damage signaling by oxidative stress in relation to DNA replication as detected using the “click chemistry” approach. Cytometry A. 2011; 79A: 897 -902. [PubMed] .
- 41. Rigby WF, Noelle RJ, Krause K, Fanger MW. The effect of 1,25-dihydroxyvitamin D3 on human T lymphocyte activation and proliferation: a cell cycle analysis. J Immunol. 1985; 135: 2279 -2286. [PubMed] .
- 42. Kowitz A, Greiner M, Thieroff-Ekerdt R. Inhibitory effect of 1alpha, 25-dihydroxyvitamin D3 on allogeneic lymphocyte stimulation and Langerhans cell maturation. Arch Dermatol Res. 1998; 290: 540 -546. [PubMed] .
- 43. Mahon BD, Wittke A, Weaver V, Cantorna MT. The targets of vitamin D depend on the differentiation and activation status of CD4 positive T cells. J Cell Biochem. 2003; 89: 922 -932. [PubMed] .
- 44. Pospelova TV, Demidenko ZN, Bukreeva EI, Gudkov VA, Blagosklonny MV. Cell Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009; 8: 4112 -4118. [PubMed] .
- 45. Burhans WC and Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007; 33: 7545 -7556. [PubMed] .
- 46. Bartkova J, Hamerlik P, Stockhausen MT, Ehrman J, Hlobikova A, Laursen H, Kalita O, Kolar Z, Paulsen HS, Broholm H, Lukas J, Bartek J. Replication stress and oxidative damage contributes to aberrant constitutive activation of DNA damage signaling in human gliomas. Oncogene. 2010; 29: 5095 -5102. [PubMed] .
- 47. Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010; 9: 3151 -3156. [PubMed] .
- 48. O'Kelly J, Uskokovic M, Lemp N, Vadgama J, Koeffler HP. Novel Gemini-vitamin D3 analog inhibits tumor cell growth and modulates the Akt/mTOR signaling pathway. J. Steroid Biochem Mol Biol. 2006; 100: 107 -116. [PubMed] .
- 49. Lisse TS, Liu M, Beckers J, Chen H, Adams JS, Hewison M. Gene targeting by vitamin D response element binding protein reveals a role for vitamin D in osteoblast mTOR signaling. FASEB J. 2011; 25: 937 -947. [PubMed] .
- 50. Lisse TS and Hewison M. Vitamin D. A new player in mTOR signaling. Cell Cycle. 2011; 10: 1888 -1889. [PubMed] .
- 51. Marcinkowska E and Kutner A. Side-chain modified vitamin D analogs require activation of both PI 3-K and erk1,2 signal transduction pathways to induce differentiation of human promyelocytic leukemia cells. Acta Biochim Pol. 2002; 49: 393 -406. [PubMed] .
- 52. Zhang Y, Zhang J, Studzinski GP. AKT pathway is activated by 1, 25-dihydroxyvitamin D3 and participates in its anti-apoptotic effect and cell cycle control in differentiating HL60 cells. Cell Cycle. 2006; 5: 447 -451. [PubMed] .
- 53. Yang J, Ikezoe T, Nishioka C, Ni L, Koeffler HP, Yokoyama A. Inhibition of mTORC1 by RAD001 (everolimus) potentiates the effects of 1,25-dihydroxyvitamin D(3) to induce growth arrest and differentiation of AML cells in vitro and in vivo. Exp Hematol. 2010; 38: 666 -676. [PubMed] .
- 54. Klotz B, Mentrup B, Regensburger M, Zeck S, Schneidereit J, Schupp N, Linded C, Merz C, Ebert R, Jakob F. 1,25-dihydroxyvitamin D3 treatment delays cellular aging in human mesenchymal stem cells while maintaining their multipotent capacity. PLoS One. 7: e29959 Epub [PubMed] .
- 55. Haussler MR, Haussler CA, Whitfield GK, Hsieh JC, Thompson PD, Barthel TK, Bartik L, Egan JB, Wu Y, Kubicek JL, Lowmiller CL, Moffet EW, Forster RE, Jurutka PW. The nuclear vitamin D receptor controls the expression of genes encoding factors which feed the “Fountain of Youth” to mediate healthful Aging. J Steroid Biochem Mol Biol. 2010; 121: 88 -97. [PubMed] .
- 56. Touhimm P. Vitamin D and aging. J Steroid Biochem Mol Biol. 2009; 114: 78 -84. [PubMed] .
- 57. Lanske B and Razzaque MS. Vitamin D and aging: old concepts and new insights. J Nutr Biochem. 2007; 18: 771 -777. [PubMed] .
- 58. Forster RE, Jurutka PW, Hsieh JC, Haussler CA, Lowmiller CL, Kaneko I, Haussler MR, Kerr WH. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochem Biophys Res Commun. 2011; 414: 557 -562. [PubMed] .
- 59. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 60. Ting H-J, Yasmin-Karim S, Yan S-J, Hsu J-W, Lin T-H, Seng W, Messing J, Sheu T-J, Bao B-Y, Li WX, Messing E, Lee Y-F. A positive feedback signaling loop between ATM and vitamin D receptor is critical for cancer chemoprevention by vitamin D. Cancer Res. 2012; 72: 958 -968. [PubMed] .
- 61. Colston K, Colston MJ, Feldman D. 1,25-dihydroxyvitamin D3 and malignant melanoma: the presence of receptors and inhibition of cell growth in culture. Endocrinology. 1981; 108: 1083 -1086. [PubMed] .
- 62. Lointier P, Wargovich MJ, Saez S, Levin B, Wildrick DM, Boman BM. The role of vitamin D3 in the proliferation of a human colon cancer cell line in vitro. Anticancer Res. 1987; 7: 817 -821. [PubMed] .
- 63. Gross M, Kost SB, Ennis B, Stumpf W, Kumar R. Effect of 1,25-dihydroxyvitamin D3 on mouse mammary tumor (GR) cells: evidence for receptors, cellular uptake, inhibition of growth and alteration in morphology at physiologic concentrations of hormone. J Bone Miner Res. 1986; 1: 457 -467. [PubMed] .
- 64. Skowronski RJ, Peehl DM, Feldman D. Vitamin D and prostate cancer: 1,25 dihydroxyvitamin D3 receptors and actions in human prostate cancer cell lines. Endocrinology. 1993; 132: 1952 -1960. [PubMed] .
- 65. Wang X, Patel R, Studzinski GP. hKSR-2, a vitamin D-regulated gene, inhibits apoptosis in arabinocytosine-treated HL60 leukemia cells. Mol Cancer Ther. 2008; 7: 2798 -2806. [PubMed] .
- 66. Gocek E, Wang X, Liu X, Liu CG, Studzinski GP. MicroRNA-32 upregulation by 1,25-dihydroxyvitamin D3 in human myeloid leukemia cells leads to Bim targeting and inhibition of AraC-induced apoptosis. Cancer Res. 2011; 71: 6230 -6239. [PubMed] .
- 67. Oka K, Tanaka T, Enoki T, Yoshimura K, Ohshima M, Kubo M, Murakami T, Gondou T, Minami Y, Takemoto Y, Harada E, Tsushimi T, Li T-S, Traganos F, Darzynkiewicz Z, Hamano K. DNA damage signaling is activated during cancer progression in human colorectal carcinoma. Cancer Biol Ther. 2010; 9: 246 -252. [PubMed] .
- 68. Studzinski GP, Wang X, Danilenko M. DNA damage response: A barrier or a path to tumor progression? Cancer Biol Ther. 2010; 9: 253 -255. [PubMed] .
- 69. Rothe G and Klouche M. Phagocyte functions. Meth Cell Biol. 2004; 75: 679 -708. .
- 70. Darzynkiewicz Z, Bedner E, Gorczyca W, Melamed MR. Laser scanning cytometry. A new instrumentation with many applications. Exp Cell Res. 1999; 249: 1 -12. [PubMed] .