



Introduction
Since its initial description cell death has appeared as a basic biological phenomenon fundamental for development and regulation of tissue homeostasis whose alteration has important implications in pathology [1]. Indeed cell death contributes to tissue homeostasis by balancing mitosis and pathology can derive from both its increase or decrease. Since the initial description of cell death in the 1960s a number of different death mechanisms have been described and have been classified both on morphological and biochemical criteria. A recent paper published in CDD [2] by a large number of experts in the field has suggested a classification of the different types and is a good reference for the subject. This review will mostly focus on the role in different human diseases of caspase dependent apoptosis Table 1).
Table 1. Diseases in which alterations of apoptosis are involved
| Cancer | |
| Breast | [20, 28, 160, 161] |
| Lung | [21, 34, 162] |
| Kidney | [22] |
| Ovary and uterus | [28] |
| CNS | [13, 23, 25, 163-165] |
| Gastro-enteric trait | [24, 30-32, 166] |
| Head and Neck | [167] |
| Melanoma | [33, 35, 168-170] |
| Lymphomas | [18, 19] |
| Leukemia | [26, 171-178] |
| Neurological disorders | |
| Alzheimer | [55, 179-183] |
| Parkinson | [63, 184, 185] |
| Huntington | [186-188] |
| Amyotrophic Lateral Sclerosis | [189-191] |
| Stroke | [192-194] |
| Cardiovascular disorders | |
| Ischemia | [99, 195-198] |
| Heart Failure | [99, 199] |
| Infectious diseases | |
| Bacterial | [200-206] |
| Viral | [150, 207-214] |
| Autoimmune diseases | |
| Systemic Lupus erythematosus | [153, 215, 216] |
| Autoimmune lymphoproliferative syndrome | [154, 155] |
| Rheumatoid arthritis | [217] |
| Thyroiditis | [1, 217, 218] |
Caspase dependent apoptosis (Figure 1) is characterized by the activation of pathways leading to the activation of a family of proteases: caspases resulting in an ordered disruption of the cell without leakage of cellular components and induction of inflammation. Apoptosis occurs following the activation of specific pathways that result in a series of well defined morphological events. The dying cell initially shows nuclear and cytoplasmic condensation, followed by blebbing of the plasma membrane that results in release of small membrane-enclosed particles containing cellular components known as apoptotic bodies. These are rapidly identified by neighbouring cells or professional phagocytes and disposed generally without induction of inflammation or tissue scarring [3]. Apoptosis depends on activation of caspases that will then cleave a number of substrates [4] resulting in the biochemical and morphological changes typical of this form of death. All caspases are synthesized as inactive zymogens that need activation to exert their function. Full activation is achieved through cleavage of a pro-domain generally by other caspases. From a functional point of view we can distinguish two types of caspases: up-stream and down-stream caspases. Up–stream caspases are activated when more enzyme molecules are brought in close proximity and undergo conformational changes upon binding to activation complexes, this results in their cleavage and full activation [5, 6]. Once activated they will activate additional molecules of the same enzyme as well as down stream caspases. Down stream caspases on the other hand can only be activated by cleavage of the pro-domain by up-stream caspase. Two main molecular pathways lead to caspase activation and therefore to apoptosis the so-called extrinsic and intrinsic pathway.
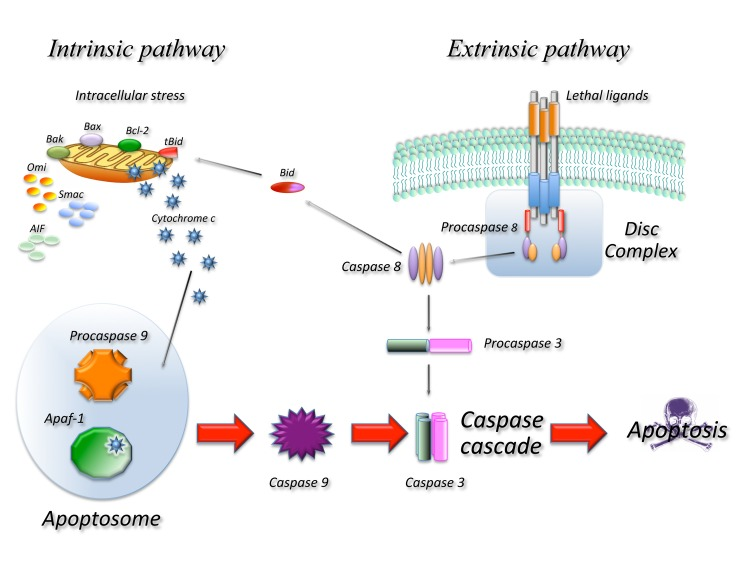
Figure 1. Schematic representation of the main molecular pathways leading to apoptosis In the extrinsic pathway upon ligand binding to specific receptors the DISC complex is formed and caspase 8 activated. In the intrinsic pathway release of cyt c from the mitochondria result in the formation of the apoptosome and activation of caspase 9. Caspase 8 and 9 then activate downstream caspases such as caspase 3 resulting in cell death. The two pathways are connected through the cleavage of the BH3 only protein BID.
Extrinsic apoptosis indicates a form of death induced by extracellular signals that result in the binding of ligands to specific trans-membrane receptors, collectively known as death receptors (DR) belonging to the TNF/NGF family. All death receptors function in a similar way: upon ligand binding several receptor molecules are brought together and undergo conformational changes allowing the assembly of a large multi-protein complex known as Death Initiation Signalling Complex (DISC) that leads to activation of the caspase cascade. In the FAS/CD95 signalling complex, that can be used as a prototype of this form of death, upon ligand binding FAS recruits, through a highly conserved 80 amino acid domain, known as death domain (DD), an adaptor molecule: Fas-associated protein with a DD (FADD). FADD contains another conserved protein interaction domain known as Death Effector Domain (DED) that binds to an homologous domain in caspase 8 leading to its activation. Active caspase 8 will activate additional caspase 8 molecules as well as downstream caspases such as caspase 3 [7].
The intrinsic pathway is activated in response to a number of stressing conditions including DNA damage, oxidative stress and many others. In all cases this multiple forms of stress converge on the mitochondria and determine mitochondrial outer membrane permeabilization (MOMP) this in turn results in dissipation of the mitochondrial membrane potential and therefore in cessation of ATP production as well as release of a number of proteins that contribute to caspase activation. At least two molecular mechanisms (not mutually exclusive) have been proposed to explain how different signals converge at the mitochondria resulting in MOMP. One involves the pore forming ability of some of the BCL-2 family proteins in the outer mitochondrial membrane [8] and the other is the result of the opening in the inner membrane of the permeability transition pore complex (PTPC), that would require the Adenine Nucleotide Transporter (ANT) and the Voltage Dependent Anion Channel (VDAC) [9, 10]. The Bcl-2 family proteins are essential regulators of this type of apoptosis and are all characterized by the presence of at least one Bcl-2 Homology (BH) domain. From a functional point of view they can be classified in anti-apoptotic members containing three or four BH domains (such as Bcl-2, Bcl-xl, Bcl-w, Mcl-1) and pro-apoptotic members with two or three BH domains (such as Bax, Bak, Bcl-xs, Bok) or with just one (such as Bad, Bik, Bid, Bim, Noxa, Puma). Pro-apoptotic members of the family mediate apoptosis by disrupting membrane integrity either directly forming pores or by binding to mitochondrial channel proteins such as VDAC or ANT, while anti-apoptotic members would prevent apoptosis by interfering with pro-apoptotic member aggregation. The different apoptotic signals are sensed by BH3 only proteins that are induced or activated and migrate to the mitochondria where they bind the pro-survival members of the family removing their block or to the pro-apoptotic members promoting their aggregation [11].
In any case once MOMP occurs a number of proteins are released from the mitochondria, these include Cytochrome C (CYTC), apoptosis-inducing factor (AIF), endonuclease G (endo G), Direct IAP-binding protein with low PI (DIABLO, also known as SMAC) and others. Once CYTC is released it binds to APAF-1 inducing the formation of a large complex, known as the apoptosome, that recruits caspase 9. In the apoptosome, caspase 9 is activated and cleaved and will activate additional molecules of caspase 9 as well as down-stream caspases such as caspase 3. Due to its lethality the system is subject to a number of controls as an example the cytoplasm contains a class of proteins known as Inhibitors of Apoptosis IAPS that bind and inactivate caspases. Upon MOMP the mitochondria also releases proteins such as DIABLO/SMAC that bind to IAPS removing their inhibition and allowing apoptosis to occur.
The intrinsic and extrinsic pathways are not completely independent: in some cells in fact activation of caspase 8 results in activation of the mitochondrial pathway. In this case caspase 8 among other things cleaves a BH3 only protein BID generating a truncated fragment known as truncated BID (tBID) that can permeabilize the mitochondrion resulting in MOMP [12].
Cancer
Based on its role in maintaining tissue homeostasis it is not surprising that alterations of apoptosis play an important role in cancer development. Moreover defects in the apoptotic pathways are responsible for resistance to therapy and new therapeutic approaches attempting to re-activate these pathways bypassing the block are currently being studied [13-17]. Alteration of many proteins involved in both intrinsic and extrinsic signaling pathways have been described and are in part summarized below. It is clear that alterations of up-stream regulators of these pathways are the most common alterations found in cancer cells, as an example p53 that can induce cell death in response to a number of different stress stimuli, is the most frequently mutated gene in human cancers. A description of these defects however is beyond the scope of this review.
BCL-2 Family proteins alterations
Bcl-2 was initially isolated and characterized in a subset of B-cell lymphomas. These tumours carry a typical translocation t(14;18) that involves Bcl-2 gene and results in its over-expression. Thus it appeared that this gene played a role in cancer even before his function in regulating apoptosis was clearly defined. Since those early days a bulk of data have proven the role of Bcl-2 alterations in development of cancer including a number of animal models. In fact transgenic mice carrying bcl-2 overexpression are actually susceptible to develop different forms of lymphoproliferative disorders [18].
Bcl-2 has been found overexpressed not only in B-cell lymphomas, but also in a variety of other cancers such as: Hodgkin lymphoma where it seems to be associated with worse overall survival [19]; breast cancer where it correlates with tumor aggressiveness, reduced survival and resistance to endocrine therapy [20]; non-small and small cell lung carcinoma (moreover squamous) [21], renal cell carcinoma [22]. A novel polymorphism of the BCL-2 promoter (-938C>A) associates with increased aggressiveness and worse prognosis in glioblastoma multiform [23], chronic lymphocytic leukemia, as well as with a better survival and outcome in breast and ovarian cancers.
In the last years a variety of alterations of the different Bcl-2 family members have been described and have proven the importance of this group of proteins in cancer development. Bax and Bak mutations [24] have been described in colon and gastric carcinomas. Unexpectedly however neither BAX single KO or BAX/BAK double KO show increased tumour formation suggesting that compensatory mechanisms can allow apoptosis in these cells. Interestingly however Bax/pARF double KO mice exhibit an increased variety of tumours (sarcomas and carcinomas). Furthermore Bax deletion accelerates growth of brain tumours in a p53 mutant mice model [25], and of mammary tumours in a SV40 large T antigen transgenic mouse model. Various BH3 protein alterations have also been implicated in cancer development, as an example Bid-deficient mice are prone to develop a form of chronic myelomonocytic leukemia [26], as well as diffuse large B-cell lymphoma.
The possibility to target Bcl-2 family members proteins to induce apoptosis in cancer cells has been studied for many years now and particular attention has been given to BH3 only proteins in designing drugs that would mimic their pro-apoptotic functions. Some of these are currently being tested in phase I/II clinical trials [27-29]. Antisense oligonucleotides targeting Bcl-2 have also been developed and in one case have reached a phase III clinical trial in patients with chronic lymphocytic leukemia.
Apoptosome defects
Apaf1 inactivation can substitute for p53 defects in promoting transformation of myc-expressing cells, moreover it is frequently silenced or inactivated in human cancers. Indeed melanomas, leukemias, glioblastomas, and cervical carcinomas have been shown to down-regulate Apaf1 by epigenetic mechanisms. In addition in some cancers a defective Cyt-C dependent caspase 9 activation in the presence of normal or elevated Apaf1 levels has been reported but the underlying molecular mechanism is still elusive and the existence of an unidentified apoptosome inhibitor has been suggested. Sequestration of Apaf1 into lipid rafts has also been demonstrated in Burkitt lymphoma cells providing another molecular mechanism for apoptosome dysfunction.
Death receptor pathway defects
Death receptor pathways alterations have profound implications in cancer and in particular in the mechanism leading to the ability of tumours to avoid immune response. In a simplistic way one can imagine a scenario in which reduced expression of death receptors protects cells from the immune system and increased surface (or soluble form) expression of death ligands allows killing of reactive cells. Indeed CD95 null mice are prone to accumulate abnormal T-cells, with splenomegaly and lymphoadenopathy and CD95 is lost or mutated in several human cancers. In fact CD95 is lost in hepatocarcinomas [30], present in less than 5% of invasive esophageal cancer cells in 79% of patients [31] where it correlates with depth of invasion and nodes metastasis, mutated in adult T cell leukemia, down-regulated in colon cancer [32], ovarian, cervical and endometrial cancers, melanoma (where lymphocyte infiltration of the invasive layer correlates with prognosis) [33] and in more than 90% of lung cancers [34]. In support of the above mentioned model, down-regulation of Fas-L leads to decreased tumor volume and increased lymphocyte infiltration. Over-expression of FAS-L has been reported in hepatocarcinomas, esophageal cancers [31], breast cancers, melanomas [35], astrocytomas , metastatic colon cancers, gastric cancers and in more than 60% of sarcomas (reaching 95% in metastatic ones). The soluble form of Fas-L (sFas-L), was also found in peripheral blood of cancer patients, where it possibly exerts an immunosuppressive effect [36, 37].
Defects of the CD95 signaling pathway can also be a consequence of alterations of other components of the DISC. FADD mutations were reported in non-small-cell lung cancer [38] and complete loss was described in a subset of diffuse large B-cell lymphoma. However FADD defects have also been shown to have a pro-oncogenic function, probably due to a role of this protein different from apoptosis. As an example phosphorylated-FADD in lung adenocarcinomas correlates with poor survival [39], and its overexpression associates with poor prognosis in oral squamous cell carcinomas.
A number of evidence highlight the importance of the Trail receptor pathway in tumour onset and development. The importance of Trail as a tumour suppressor is supported by a number of results in different animal models including the onset of spontaneous haematopoietic malignancies in Trail KO mice [40]. As described for CD95 also in the case of Trail, defects mostly allow tumour cells to escape immune control. Trail receptors 1 and 2 map to chromosome 8p21-22 frequently lost in tumours and mutations of trail receptors have been described in up to 20% of various human tumours, including breast cancer, head and neck cancers, and non-Hodgkin lymphomas.
The possibility to develop cancer therapies based on the activation of death receptors has been attractive since their discovery, however toxicity of therapies targeting TNF and CD95 have greatly reduced the initial enthusiasm. The finding that recombinant Trail preferentially induced apoptosis in cancer cells while sparing normal cells supported by its low or absent toxicity in vivo [40, 41] have attracted growing interest on the possibility to exploit this pathway for cancer therapy. Despite a large investigative effort however the reason for this selective activity is poorly understood.
Altered caspase activity
Caspases are the final effectors of both extrinsic and intrinsic apoptosis, therefore it is expected that interfering with their function impairs these pathway leading to a survival advantage for cancer cells and indeed caspase alterations are not rare in a variety of tumours. These can be due to mutations, promoter methylation, alternative splicing, and post-translational modifications. Some of these defects are loss of functions, but in other cases mutated caspases act as dominant negatives preventing activation also of the wild type protein [42].
Altered caspase function can also be a consequence of modified expression of their specific inhibitors, as an example cFLIPs that competes with caspase 8 for FADD binding, thus preventing its activation, is often elevated in tumours, while its down-regulation can sensitize tumour cells to therapy. Among caspase inhibitors an important role is played by IAPs. Indeed alterations of IAPs are found in a variety of human cancers and are associated with poor prognosis and resistance to therapy. In some cases however loss of IAPs correlates with tumour progression complicating the issue and suggesting that the role of IAPs has to be carefully evaluated based on cell context. While initially described as caspase inhibitors now IAPs have been recognized to regulate a multitude of other cellular functions including regulation of the immune response cell migration, mitosis and proliferation [43]. As many of these processes are often modified in cancer it is clear how alteration of IAPs can play a role in tumorigenesis not only as a consequence of altered apoptosis. In fact probably the most important pathway regulated by IAPs that contributes to cancer development is the NF-kB signaling pathway. XIAP, cIAP1 and cIAP2 have been shown to regulate this pathway and as a consequence inflammation, immunity and cell survival. Moreover cIAPs protect from TNF killing. In addition, recent findings show a role for IAPs in metastatization as a XIAP/survivin complex would trigger NF-kB pathway leading to activation of cell motility kinases [43]. This however is still a controversial issue and other studies show a suppressive effect of IAPs on cell mobility.
In any case due to their involvement in cancer progression and to their ability to suppress apoptosis IAPs have become an attractive therapeutical target, leading to the development of IAP inhibitors, some of which are based on natural inhibitors such as Smac/DIABLO [43, 44]. These drugs appear to be able to directly kill cancer cells or at least sensitize them to other killing agents while sparing normal cells. A number of these compounds are currently entering clinical trials and their efficacy will be evaluated in the next few years.
Neurological disorders
From a physiological point of view apoptosis plays a key role in central nervous development [45, 46], while in adult brain it is involved in the pathogenesis of a number of diseases including neurodegenerative diseases and acute injury (i.e. stroke).
Neurodegenerative diseases
Alzheimer's disease (AD) is the seventh leading cause of all deaths in the United States, it is a progressive neurodegenerative disorder characterized by accumulation of amyloid-β β peptides in extracellular senile plaques intra-cellular neurofibrillary tangles (NFTs) formation resulting from hyper-phosphorylated microtubule-associated protein tau resulting in loss of neurons and consequent progressive dementia [47-49].
Neuronal apoptosis plays an important role in AD pathogenesis and caspases seem to be involved also in some of the upstream pathological events. Exposure of cultured hippocampal neurons to β results in caspase 3 activation and apoptosis [50]. Aβ is generated following sequential cleavage of the amyloid precursor protein (APP), and caspase 3 is considered the predominant caspase involved in APP cleavage [51, 52]. Tau protein is also a substrate for caspase 3; cleavage of tau at its C-terminus would promote tau hyper-phosphorylation and accumulation of NFTs. Moreover, β-induced caspase 3 activation causes abnormal processing of the tau protein in models of AD [53]. APP is also cleaved by caspase 6 in vivo [54], moreover the N-terminal APP fragment is a ligand for death receptor 6 (DR6 also known as TNFRSF21) activation of which triggers caspase 6 dependent axonal degeneration [55].
The potential benefit of inhibiting the intrinsic apoptotic pathway has been suggested through the use of a triple transgenic AD mouse model wherein overexpression of the anti-apoptotic Bcl-2 gene blocked activation of caspases 9 and 3; in these conditions, the degree of caspase cleavage of tau was limited, the formation of plaques and tangles was inhibited, and memory retention was improved [56, 57].
Parkinson's disease (PD) is considered the 2nd most common chronic neurodegenerative disorder after AD, it is associated with movement disorders, tremors, and rigidity and is characterized by a specific loss of dopaminergic neurons of the substantia nigra. This degeneration leads to the formation of fibrillar cytoplasmic inclusions known as Lewy bodies. A preponderant role of the aberrant activation of intrinsic and extrinsic apoptotic pathways in PD pathogenesis has been suggested. The involvement of caspases 1 and 3 in apoptotic cell death has been proved using PD animal models [58]. PD has been linked to mutations in several genes such as parkin [59], DJ-1, and a gene codifying for a mitochondrial kinase, (PTEN)-induced kinase 1 (PINK1) [60]. PINK1 function is related to the inhibition of mitochondria-dependent apoptosis [61]. In human and mouse neurons deleted for PINK1 Bax translocation to the mitochondria and cytochrome c release to the cytoplasm occur earlier than in control cells. Furthermore loss of PINK1 results in elevated levels of caspase activation (caspases 3 and 9) [61]. Gene-expression profiling studies performed on material from patients affected by PD confirmed down-regulation of PINK1 as well as other anti-apoptotic proteins such as Bcl-2 but also found evidence for the involvement of the extrinsic pathway. Indeed death receptors such as FAS, TNFRSF10B and TNFRSF21 were up-regulated in PD-affected neurons [62, 63].
Huntington's disease (HD) is a disorder characterized by a degenerative process, which affects medium spiny striatal and cortical neurons. HD is an autosomal dominant disease caused by a mutation in the gene encoding the huntingtin protein (htt); this mutation is responsible for abnormal expansion of a trinucleotide CAG repeat encoding polyglutamine tract expansion in the N terminus of htt [64]. The expanded polyglutamine alters protein folding, leading to generation of aggregates in neurons that seem to be crucial for the neurodegenerative process [65, 66]. Mutant htt is cleaved by different proteases including caspases [67] and accumulation of caspase cleaved fragments is an early pathological finding in brains of HD patients [68]. Moreover transgenic mice models have demonstrated that caspase 6 cleavage of mutant htt is required for the development of the characteristic behavioral and neuro-pathological symptoms. In addition activation of caspase 6, is observed before the onset of motor abnormalities in HD brains, suggesting that these activation could be used as an early marker of the disease [69].
An additional molecular mechanism involves htt-interacting protein 1 (HIP-1) that binds a polypeptide named Hippi (HIP-1 protein interactor) forming a complex that can activate caspase 8. The free cellular HIP-1 concentration is increased when htt is mutated (HD), this would favor the pro-apoptotic Hippi-Hip complex formation [70, 71].
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by muscle atrophy, paralysis, and, death due to progressive loss of motor neurons [72]. About 10% of cases are familial as a result of mutations in the copper-zinc superoxide dismutase (SOD1) gene [73], whereas the majority of them are sporadic. SOD1 catalyzes conversion of the superoxide anion to hydrogen peroxide, however the mechanism by which SOD1 mutations cause ALS is still not completely understood. Several alterations have been identified in ALS and are thought to play a role in motor neuron death, including: neurofilament abnormalities, aggregate formation, oxidative stress, and inflammatory processes [74]. Mice overexpressing a human mutant SOD1 develop neuron degeneration, and have been used as a model [75, 76]. These mice show increased p38 activity that determines increased NO production that in turn results in increased FasL expression and activation of the extrinsic pathway [77]. In addition mutated SOD would localize to the mitochondria and directly trigger CYTC release and therefore neuronal death [78].
Motor neurons degeneration in ALS is also accompanied by inflammation, but the exact mechanism triggering inflammatory response remains unclear. Meissner et al have recently reported that mutant SOD1 induces IL-1 beta and promotes caspase 1 activation resulting in neuro-inflammation that would contribute to the pathogenesis [79]. Finally caspase 1 would act as a chronic activator of caspase 3 contributing to neuronal loss [80].
Acute CNS insults
Stroke is the leading cause of acquired adult disability in USA [81]. Ischemic injury is caused by the loss of blood flow to the brain, usually as a consequence of an embolism. The decrease in perfusion determines both apoptotic and necrotic neuronal cell death in the affected region (core) due to energy depletion [82, 83]. Around this area of tissues that is irreversibly lost there is an area of partially damaged tissue known as penumbra that triggers local inflammation. Several evidences suggest that inflammation is a crucial event in the progression of ischemic brain damage. Cyclooxygenase-2 (COX-2) induction and prostaglandin E2 elevation have been reported to occur after cerebral ischemic insult [84]. Takadera et al showed that prostaglandin E2 directly induced apoptosis in hippocampal neurons through the activation of caspase 3 [85], suggesting that a direct effect of prostaglandin E2 on hippocampal neurons was mediated by activation of the EP2 receptors. The deletion of EP3 receptors is known to ameliorate stroke injury in experimental stroke models [86], and recently it has been demonstrated that EP3 receptors are involved in the enhancement of inflammatory and apoptotic response in the ischemic cortex [87].
An important role in ischemic brain damage seems to be played also by activation of the receptor pathway. TNF deletion in mice protects the brain from ischemic damage [88]. Fas and FasL levels seem to be increased during brain ischemia [89], and interfering with the Fas signaling pathway using a blocking anti-FasL antibody markedly reduces death of neurons and improves functional recovery in animal models of stroke and spinal injury. Moreover chronic extrinsic cervical spinal cord compression leads to Fas-mediated apoptosis of neurons [90].
C-Jun N-terminal protein kinase (JNK) signaling pathway is known to be activated in response to stress and ischemia. JNK activation precedes inflammation and apoptosis in neuronal cells [91]. Indeed, JNK is involved in the regulation of several pro-apoptotic proteins such as Bim and inhibition of JNK activity attenuated Bax translocation in ischemic neurons [92].
The involvement of caspases in mediating ischemic neuronal cell death has been also demonstrated using caspase 1 and 3 KO mice. Cortical neurons from caspase 3 -/- mice subjected to oxygen-glucose deprivation, were more resistant to cell death [93], while caspase 1 deletion in mice led to reduced production of IL-1 beta [94]. Consistently the reduction of IL-1 beta function, using specific antagonists, results in neuroprotective effects in stroke animal models [95].
Benchoua et al. proposed activation of different and specific pathways in the core or in the penumbra area of the brain infarction. In particular, after cerebral infarction, in neurons of the core area the first apoptotic events are mediated by ligand binding to specific death receptors leading to caspase 8 activation. In the penumbral area, where mitochondria provide residual energy supply, neuronal death is instead induced through the mitochondrial pathway [96].
The reperfusion of ischemic tissue usually improves clinical outcome of patients, but in others it may amplify brain damage due to the ischemia-reperfusion injury. Reactive oxygen species (ROS) levels are immediately increased after a vessel occlusion is cleared, and are considered the main mediators of reperfusion injury [97] resulting in the release of cytochrome C [98].
Heart diseases
Adult cardiomyocites are post-mitotic cells, therefore this tissue has limited response capability to damage. In general acute damage results in cell death of various types while chronic stress mainly results in hypertrophy and myocardial remodelling. Increased evidences suggest that slow turnover exists in the normal myocardium sustained by stem cells. In pathological conditions however death exceeds mitosis resulting in heart failure. Apoptosis is very rare in normal myocardium with a reported rate of 0.001-0.002% however it is increased in both acute and chronic heart pathologies were it seems to play an important role [99].
Ischemia reperfusion
Occlusion of coronary arteries results in myocardial infarction characterized by massive cell death due to deprivation of oxygen, nutrients and survival factors. Reperfusion of the ischemic tissue is the treatment for acute coronary syndromes and limits the size of the lesion but still results in damage due to oxidative stress, increased cytosolic and mitochondrial calcium levels and inflammation determining what is known as reperfusion injury. Apoptosis plays an essential role in the pathogenesis of I/R and in general prolonged ischemia determines an increase in necrosis while reperfusion leads to increased apoptosis. Death during ischemia/reperfusion (I/R) is of different kinds: necrotic apoptotic and autophagic. Both the intrinsic and extrinsic apoptotic pathway are involved in I/R. Indeed lpr mice (FAS deficient) show reduced infarct size while deletion of both TNFR1 and TNFR2 appears to have a protective effect. The role of the intrinsic pathway is demonstrated by reduced apoptosis in cardiomiocytes over-expressing Bcl-2 or lacking Bax, both in vitro and in vivo. As explained above the two pathways are connected through Bid cleavage that is indeed observed in I/R and Bid KO mice show reduced infarct size following I/R. Similarly deletion of PUMA (a BH3 only protein activated by p53) also protects from apoptosis suggesting the involvement of the p53 pathway [99]. A number of studies have shown that inhibitors of caspases can protect cardiomyocites reducing the infarct size. Interestingly over-expression of constitutively active Akt or pharmacological sustained activation, shows significant protection from apoptosis probably by inactivating a number of pro-apoptotic proteins [100].
Ischemia damages several mitochondrial components including proteins of the oxidative phosphorylation complexes and membranes and predisposes them to ROS generation [101]. Indeed it is well known that the production of ROS is greatly increased during the reperfusion phase when oxygen becomes available and the mitochondrial respiratory chain is impaired. Furthermore this is exacerbated by reduced antioxidant defenses.
Chronic Heart Failure
Heart failure is a complex syndrome where the heart is incapable of o meeting the metabolic requirement of the body, it can be the result of a number of other heart diseases such as myocardial infarction, cardio-myopathies and hypertension. In all cases it is characterized by left ventricular remodeling with chamber enlargement and wall thinning. During heart failure cardiomiocytes show a modest increase in apoptosis, that however seems to play an important role in the pathogenesis [99]. The estimated rate of apoptosis in cardiomiocytes in patients with dilated cardiomyopathy is between 0.08% and 0.25% and increasing apoptosis by only 0.023% in cardiomyocites by over-expression of caspase 8 in mice results in dilated cardiomyopathy after 2-6 months, that is reduced by treatment with caspase inhibitors [102]. Multiple stress stimuli have been implicated in activating apoptosis in cardiomyocites eventually leading to heart failure, these include: excessive mechanical stress, ROS, β1 adrenergic receptor agonists, angiotensin II, cytokines. The ASK 1-JNK pathway seems to play an important role in mediating ROS dependent apoptosis in these cells (in part trough inactivation of Bcl-2) and regulating left ventricular remodeling. Indeed it has been proposed that inhibition of Ask-1 can attenuate heart failure [103]. Another important pathway seems to involve Gαq, that transduces the signal of a number of receptors including angiotensin II receptor and α1-adrenergic receptor. Over-expression of Gαq in mice heart results in heart failure, this is accompanied by over expression of Nix/Bnip3L, a BH3 like protein, that can induce cardiomiocyte apoptosis [99].
Infectious diseases
Pathogenic microorganisms, once inside a host must avoid detection and destruction for as long as possible. Several pathogens are able to trigger or inhibit apoptosis in eukaryotic host cells thus escaping the immune system. On the other hand the host can use apoptosis in an attempt to defend himself from pathogens. Thus, apoptosis has a fundamental role in cellular host-pathogen interactions that depends on the nature of the pathogen, the cell type and the intensity of the infection.
Bacterial infections
Although it is outside the scope of this review, it is worth mentioning a physiological role for apoptosis in bacteria [104]. While initially counterintuitive, several studies have underlined how this process may be fundamental to bacterial physiology. As an example, autolysis, the most common known form of apoptosis in bacterial cells, is used by Bacillus subtilis during sporulation to eliminate a barrier, the cell wall that could interfere with spore germination. Also, when exposed to harmful conditions as antimicrobial agents, to eliminate defective cells, damaged bacteria self-digest the cell wall by peptidoglycan hydrolases. Moreover, bacterial apoptosis maintains biofilm homeostasis by balancing cell death and viability. The increasing knowledge of ancestral forms of apoptosis in bacteria will possibly shed light on the evolution and molecular mechanisms of apoptosis in eukaryotic cells. More relevantly to the subject of this review it is well known that bacteria can modulate apoptosis in the host favoring their survival and propagation. Several apoptogenic molecules have been identified in bacteria including common structural components as lipopolysaccharide in Gram-negatives [105], lipotechoic acid the major constituent of the cell wall of Gram-positives and lipoarabinomannan in Mycobacteria [106] as well as virulence factors such as exotoxins [107], cytolysins [108] and hemolysins [109]. Through all these different molecules, bacteria are able to trigger both intrinsic and extrinsic apoptotic pathways, here we report some examples in different types of both endo-cellular and extracellular bacteria.
Mycobacterium tuberculosis continues to be a major cause of pulmonary infection in the world. It is an intracellular parasite capable of establishing a long-term infection. It lacks toxins and exerts its virulence through its cell wall components [110]. The initial infection typically occurs in the alveolar spaces of the lung where bacteria are phagocytized by macrophages. In the alveolar macrophages, bacteria can survive and replicate by preventing fusion of phagosomes with lysosomes and thus their cellular lysis [110]. The apoptotic response to intracellular pathogens may favour both host and parasite in several ways. The host responds to the presence of intracellular M. tuberculosis triggering a TNF-α-mediated apoptotic pathway. On the contrary, bacteria block the TNF-α signalling by up-regulating the anti-apoptotic protein Mcl-1 [111]. Recently it has been reported that mycobacteria may trigger a non-classical type of apoptosis to exit host cells [112]. Cell death is induced when a threshold of about 20 bacteria per macrophage is reached and begins with some typical features of apoptosis followed by secondary necrosis and release of bacteria. To indicate this non-classical mode of apoptosis, Lee and colleagues coined the expression “high-MOI apoptosis” [111]. In addition Mycobacteria secrete a 19kDa lipoprotein that binds the TLR2 receptor that signals through myeloid differentiation factor 88 (MyD88) activating apoptosis in macrophages [113].
Pseudomonas aeruginosa is an opportunistic, multidrug resistant bacterium that often causes severe nosocomial infections such as urinary tract infections, pneumonia and bacteremia [114]. P. aeruginosa is also responsible for pulmonary infection of patients with cystic fibrosis [114]. Although considered as an extracellular pathogen, P. aeruginosa can invade and survive within different types of cells, in particular epithelial respiratory cells. P. aeruginosa triggers cell death of lung epithelial cells through the CD95 pathway [115]. In vivo experiments have demonstrated that, in this case, apoptosis plays a role in the host defence against the bacterial infection [115]. Usually, P. aeruginosa is aspired into the lower respiratory tract after colonizing the oral cavity where oral bacteria may facilitate the adhesion and invasion of P. aeruginosa into respiratory epithelial cells followed by cytokine release and apoptosis [116]. Indeed co-incubation with oral bacteria resulted in increased induction of apoptosis in host cells [116].
Chlamydiae are obligate intracellular bacteria that have adapted to endo-cellular life to the extent of no longer being able to replicate outside the host. Chlamydia trachomatis is a common sexually transmitted human pathogen that causes urogenital infections, conjunctivitis and trachoma, while Chlamydophila pneumoniae is commonly associated with upper respiratory tract infections and can give rise to community-acquired pneumonia [117]. Apoptosis-inhibiting mechanism have been shown in both and appear to be similar [105]. During persistent infection, Chlamidiae inhibit the release of cytochrome c thus blocking apoptosis [118]. The underlying molecular mechanism seems to be the proteolytic degradation of BH3-only proteins by a chlamydial protease-like activity factor (CPAF) [119].
Enteric pathogens producing Shiga toxins Shiga toxins (Stxs) consist of a family of related cytotoxic proteins expressed by the enteric pathogen Shigella dysenteriae serotype 1 and by several serotypes of Enterohaemorrhagic Escherichia coli (EHEC; Shiga toxin-producing E. coli, STEC), Shigellae are endocellular human-adapted pathogens transmitted by the fecal-oral route. STEC are frequently associated with severe disease, while not invasive they are able to adhere to the intestinal epithelium, causing severe cell alterations. Stxs inhibit protein synthesis by inactivation of eukaryotic ribosomes [120], they have an AB5 structure consisting of an enzymatically active A-subunit and a pentameric B-subunit that bind the toxin receptor globotriaosylceramide (Gb3), a neutral glycolipid of the globo-series. Stxs are transported in a retrograde manner, through the Golgi, to the endoplasmic reticulum allowing the transfer of the A-subunit into the cytosol where it inhibits protein synthesis [121].
Shigellae and EHEC can survive phagocytosis by inducing apoptosis through activation of the MAPK pathway [107, 122]. Moreover through the prolonged activation of the ribotoxic and endoplasmic reticulum stress responses, they may initiate apoptosis with the rapid activation of caspase 8 and activation of both intrinsic and extrinsic pathways [107].
Interestingly it has also been demonstrated that Stxs induce apoptosis in cancer cells and therefore could be used as potential anticancer agents [123]. However, an obstacle is constituted by the possible toxicity since they can also inhibit protein synthesis and trigger apoptosis in normal cells.
Helicobacter pylori is the major cause of gastroduodenal diseases including peptic ulcer disease, gastric lymphoma and gastric adenocarcinoma [124]. Vacuolating cytotoxin (VacA) is one of several virulence factors identified in H. pylori [124]. In addition to cell vacuolation, VacA has other effects on different cell types, including membrane channel formation, inhibition of T-cell activation and proliferation as well as apoptosis. It has been documented that VacA induces apoptosis through the mitochondrial-dependent pathway in gastric epithelial cells [125]. The toxin binds to the RPTPβ (receptor-like protein tyrosine phosphatase) receptor and activates the pro-apoptotic Bcl-2 family proteins, Bax and Bak. In addition, VacA causes down-regulation of JAK-STAT3 signaling pathway resulting in reduced expression of the anti-apoptotic Bcl-2 family proteins, Bcl-2 and Bcl-XL.
Staphylococcus aureus is an ubiquitous bacterium and is a part of the human skin and mucosal flora, it can engender a variety of diseases and it is one of the most common causes of hospital infections. It secretes several virulence factors, exotoxins and enzymes, involved in the pathogenic mechanisms of the bacterium. Two virulence factors have mainly been involved in induction of cell death. Superantigen enterotoxin B directly binds to both MCH class II molecules and to the variable regions of the T-cell receptor beta chain (TCRVbeta), cross-linking them in a non-specific way, resulting in polyclonal T-cell activation which can be followed by cell death [126]. Enterotoxin B causes activation-induced cell death of T lymphocytes, a process involving TCR binding, activation, cell expansion, FAS expression and finally PCD. Staphylococcal α-toxin is an hemolysin that causes pore formation in the host cell membranes, and the consequent disruption of the Na+/K+ balance appears to trigger apoptosis through regulation of Bcl-2 and CYTC release.
Streptococcus pneumoniae is part of the human respiratory tract normal flora and is the common cause of community-acquired pneumonia, bacterial meningitis and bacteremia. Although the virulence of this pathogen is in large part determined by its capsular polysaccharide, it produces several additional virulence factors involved in the infection mechanisms. Pneumolysin, like α-toxin as an example, is a pore forming exotoxin that can trigger cell death in macrophages and other cell types. In human macrophages it permeabilizes mitochondrial membranes causing cytochrome c release [127].
Bacillus anthracis is the causative agent of anthrax a well known zoonotic disease. Three different primary forms of the disease are recognized, the cutaneous, the inhalation and the gastrointestinal form. The bacterium has two major virulence factors, the poly-D-glutamate capsular filaments that plays a major role as an invasiveness factor and a toxin complex [128]. The toxin complex is a tripartite exotoxin that consists of three polypeptide subunits: the protective antigen, the edema factor and the lethal factor. The protective antigen binds to cellular receptors and mediates the entry into the cytosol of the other two factors. The lethal factor is a Zn+ metalloprotease that inhibits the MAPK pathway causing cell death, while the edema factor is an adenylate cyclase that converts ATP to cyclic AMP and promotes lethal tissue edema [128]. Both lethal factor and edema factor inhibit acquired and innate immune responses, allowing the bacteria to multiply in the host.
Listeria monocytogenes is involved in a foodborne illness characterized by gastroenteritis, meningitis, encephalitis and sepsis [129]. It is a facultative endo-cellular bacterium that grows in macrophages, epithelial cells and fibroblasts [129] and can induce apoptosis in several cell types [130]. In hepatocytes, activation of apoptosis probably contributes to the resolution of infection by: (i) elimination of infected cells and reduction of host cells for bacterial replication and (ii) allowing access of immune cells to the microorganisms. On the contrary, Listeriae can induce cell death to reduce the number of phagocytes, lowering antigen presentation and breaking efficient adaptative immune response. The hemolysin listeriolysin O, the most widely studied virulence factor of L. monocytogenes promotes apoptosis in host T cells [108], through a mechanism that involves loss of mitochondrial membrane potential [108].
Clostridium difficile causes nosocomial diarrhoea in adults by colonizing the lower intestinal tract [131]. Two virulence factors, enterotoxins A and B, largely responsible for pathogenicity, are both inducers of apoptosis. In intestinal epithelial cells, the toxins produce a loss of mitochondrial membrane potential followed by release of cytochrome C [130]. The decrease of Rho protein activity appears necessary to trigger apoptosis [132]. These toxins can also trigger the extrinsic pathway [130].
Autoimmune diseases
A common feature of autoimmune diseases is altered tolerance to self antigens and generation of autoantibodies. Immune homeostasis and maintenance of immune tolerance are strongly dependent on apoptosis, moreover defective clearance of dying cells results in persistence of autoantigens, therefore autoimmune diseases can arise both from defective clearance of autoreactive cells or by delayed elimination of autoantigens. In addition increased apoptosis as a consequence of viral infections, gamma irradiation or other stressing conditions may contribute to disease onset. More recently it has been suggested that upon apoptosis and/or secondary necrosis autoantigens are cleaved and modified exposing novel epitopes that are recognized by the immune system, again altered or delayed clearance as well as prolonged exposure to apoptotic inducing stimuli would result in autoimmune response. Moreover formation of immune complexes would result in secretion of pro-inflammatory cytokines such as IL-8, Il-1β, TNFβ and IFN-α resulting in chronic inflammation and organ damage.
A large number of evidence support the idea that defective apoptosis of immune cells leads to autoimmune disease. Lpr and gld mice defective for the Fas signaling pathway develop lymphoadenopathy and splenomegaly and produce a large number of autoantibodies developing a disease that resembles human systemic lupus erythematosus (SLE), clearly demonstrating an essential role for the extrinsic apoptotic pathway in controlling autoreactive T and B cells and the fact that alteration in apoptosis can strongly contribute to autoimmune diseases pathogenesis [152, 153]. In humans defects of the Fas signaling pathway lead to the autoimmune lymphoproliferative syndrome (ALPS) characterized by non-malignant lymphoproliferation and autoimmunity and have increased incidence of malignancies. 70% of these patients carry germ-line heterozygous FAS mutations, while the rest have somatic FAS mutations or mutations of FAS ligand, caspase 10 and caspase 8. In most cases mutations function as dominant negatives inhibiting also the function of the wild type protein [154].
While defects in the extrinsic pathway seem to play a major role in the immune system it is becoming clear that also the intrinsic pathway participates and its alterations can contribute to autoimmune disease pathogenesis [155]. As an example Bim KO mice have been shown to accumulate lymphoid and myeloid cells and develop an autoimmune disease, [156, 157]. While no mutations of the BH3 only proteins have been described in patients with autoimmune diseases, reduced levels of Bim were reported in a patient with ALPS and over-expression of pro-survival members of the bcl2 family have been reported in SLE [155].
As mentioned above altered clearance of apoptotic cells also contributes to autoimmune diseases pathogenesis. In fact MGF-8 (a protein essential for macrophage clearance of apoptotic cells) defective mice also produce a large number of autoantibodies and develop a SLE-type autoimmune disease [152]. This is probably due to release of cellular material from apoptotic cells that have not been cleared by phagocytosis and undergo secondary necrosis. These cellular antigens would activate autoimmunity. Along the same line mice defective for the complement component C1q develop a SLE-like glomerulonephritis. Moreover defective clearance of apoptotic cells has been demonstrated in patients with SLE [158] and macrophages from at least 50% of these patients show reduced phagocytosis [159].
Concluding remarks
Over 50 years of research in the field of cell death have clarified many aspects of this fundamental process and brought to the attention of scientist its role in a large number of different diseases, however exploitation of this knowledge in therapy is only at its early steps. We expect that in the following years more approaches based on control of the different forms of cell death will enter the clinical practice. Many problems need to be solved, of course, such as activation of alternative death pathways when one is pharmaceutically blocked, in diseases where survival of the target cell is the final goal, as well as unwanted death of “innocent bystanders”, when attempting to kill pathological cells such as in anti-tumor therapies.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Knight RA and Melino G. Cell death in disease: from 2010 onwards. Cell Death Dis. 2011; 2: e202 [PubMed] .
- 2. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2011; .
- 3. Wickman G, Julian L, Olson MF. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012; 19: 735 -742. [PubMed] .
- 4. Ai X, Butts B, Vora K, Li W, Tache-Talmadge C, Fridman A, Mehmet H. Generation and characterization of antibodies specific for caspase-cleaved neo-epitopes: a novel approach. Cell Death Dis. 2011; 2: e205 [PubMed] .
- 5. Luthi AU and Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 2007; 14: 641 -650. [PubMed] .
- 6. Tadokoro D, Takahama S, Shimizu K, Hayashi S, Endo Y, Sawasaki T. Characterization of a caspase-3-substrate kinome using an N- and C-terminally tagged protein kinase library produced by a cell-free system. Cell Death Dis. 2010; 1: e89 [PubMed] .
- 7. Lavrik IN and Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012; 19: 36 -41. [PubMed] .
- 8. Gavathiotis E and Walensky LD. Tracking BAX once its trigger is pulled. Cell Cycle. 2011; 10: 868 -870. [PubMed] .
- 9. Green DR and Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004; 305: 626 -629. [PubMed] .
- 10. Yivgi-Ohana N, Eifer M, Addadi Y, Neeman M, Gross A. Utilizing mitochondrial events as biomarkers for imaging apoptosis. Cell Death Dis. 2011; 2: e166 [PubMed] .
- 11. Chipuk JE and Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008; 18: 157 -164. [PubMed] .
- 12. Kaufmann T, Strasser A, Jost PJ. Fas death receptor signalling: roles of Bid and XIAP. Cell Death Differ. 2012; 19: 42 -50. [PubMed] .
- 13. Goldberg AA, Beach A, Davies GF, Harkness TA, Leblanc A, Titorenko VI. Lithocholic bile acid selectively kills neuroblastoma cells, while sparing normal neuronal cells. Oncotarget. 2011; 2: 761 -782. [PubMed] .
- 14. Eberhard Y, Gronda M, Hurren R, Datti A, MacLean N, Ketela T, Moffat J, Wrana JL, Schimmer AD. Inhibition of SREBP1 sensitizes cells to death ligands. Oncotarget. 2011; 2: 186 -196. [PubMed] .
- 15. Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget. 2011; 2: 209 -221. [PubMed] .
- 16. Burz C, Berindan-Neagoe I, Balacescu O, Irimie A. Apoptosis in cancer: key molecular signaling pathways and therapy targets. Acta Oncol. 2009; 48: 811 -821. [PubMed] .
- 17. Jazirehi AR. Regulation of apoptosis-associated genes by histone deacetylase inhibitors: implications in cancer therapy. Anticancer Drugs. 2010; 21: 805 -813. [PubMed] .
- 18. Egle A, Harris AW, Bath ML, O'Reilly L, Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood. 2004; 103: 2276 -2283. [PubMed] .
- 19. Gascoyne RD, Adomat SA, Krajewski S, Krajewska M, Horsman DE, Tolcher AW, O'Reilly SE, Hoskins P, Coldman AJ, Reed JC, Connors JM. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin's lymphoma. Blood. 1997; 90: 244 -251. [PubMed] .
- 20. Tawfik K, Kimler BF, Davis MK, Fan F, Tawfik O. Prognostic significance of Bcl-2 in invasive mammary carcinomas: a comparative clinicopathologic study between ‘triple-negative’ and non-‘triple-negative’ tumors. Hum Pathol. 2011; .
- 21. Han H, Landreneau RJ, Santucci TS, Tung MY, Macherey RS, Shackney SE, Sturgis CD, Raab SS, Silverman JF. Prognostic value of immunohistochemical expressions of p53, HER-2/neu, and bcl-2 in stage I non-small-cell lung cancer. Hum Pathol. 2002; 33: 105 -110. [PubMed] .
- 22. Gobe G, Rubin M, Williams G, Sawczuk I, Buttyan R. Apoptosis and expression of Bcl-2, Bcl-XL, and Bax in renal cell carcinomas. Cancer Invest. 2002; 20: 324 -332. [PubMed] .
- 23. El Hindy N, Bachmann HS, Lambertz N, Adamzik M, Nuckel H, Worm K, Zhu Y, Sure U, Siffert W, Sandalcioglu IE. Association of the CC genotype of the regulatory BCL2 promoter polymorphism (-938C>A) with better 2-year survival in patients with glioblastoma multiforme. J Neurosurg. 2011; 114: 1631 -1639. [PubMed] .
- 24. Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997; 275: 967 -969. [PubMed] .
- 25. Yin C, Knudson CM, Korsmeyer SJ, Van Dyke T. Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature. 1997; 385: 637 -640. [PubMed] .
- 26. Zinkel SS, Hurov KE, Ong C, Abtahi FM, Gross A, Korsmeyer SJ. A role for proapoptotic BID in the DNA-damage response. Cell. 2005; 122: 579 -591. [PubMed] .
- 27. Esposti MD. Bcl-2 antagonists and cancer: from the clinic, back to the bench. Cell Death Dis. 2010; 1: e37 [PubMed] .
- 28. Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010; 1: e40 [PubMed] .
- 29. Kelly PN and Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011; 18: 1414 -1424. [PubMed] .
- 30. Strand S, Hofmann WJ, Hug H, Muller M, Otto G, Strand D, Mariani SM, Stremmel W, Krammer PH, Galle PR. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells--a mechanism of immune evasion? Nat Med. 1996; 2: 1361 -1366. [PubMed] .
- 31. Gratas C, Tohma Y, Barnas C, Taniere P, Hainaut P, Ohgaki H. Up-regulation of Fas (APO-1/CD95) ligand and down-regulation of Fas expression in human esophageal cancer. Cancer Res. 1998; 58: 2057 -2062. [PubMed] .
- 32. Butler LM, Hewett PJ, Butler WJ, Cowled PA. Down-regulation of Fas gene expression in colon cancer is not a result of allelic loss or gene rearrangement. Br J Cancer. 1998; 77: 1454 -1459. [PubMed] .
- 33. Bullani RR, Wehrli P, Viard-Leveugle I, Rimoldi D, Cerottini JC, Saurat JH, Tschopp J, French LE. Frequent downregulation of Fas (CD95) expression and function in melanoma. Melanoma Res. 2002; 12: 263 -270. [PubMed] .
- 34. Viard-Leveugle I, Veyrenc S, French LE, Brambilla C, Brambilla E. Frequent loss of Fas expression and function in human lung tumours with overexpression of FasL in small cell lung carcinoma. J Pathol. 2003; 201: 268 -277. [PubMed] .
- 35. Hahne M, Rimoldi D, Schroter M, Romero P, Schreier M, French LE, Schneider P, Bornand T, Fontana A, Lienard D, Cerottini J, Tschopp J. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996; 274: 1363 -1366. [PubMed] .
- 36. Ueno T, Toi M, Tominaga T. Circulating soluble Fas concentration in breast cancer patients. Clin Cancer Res. 1999; 5: 3529 -3533. [PubMed] .
- 37. Kavathia N, Jain A, Walston J, Beamer BA, Fedarko NS. Serum markers of apoptosis decrease with age and cancer stage. Aging (Albany NY). 2009; 1: 652 -663. [PubMed] .
- 38. Shin MS, Kim HS, Lee SH, Lee JW, Song YH, Kim YS, Park WS, Kim SY, Lee SN, Park JY, Lee JH, Xiao W, Jo KH, et al. Alterations of Fas-pathway genes associated with nodal metastasis in non-small cell lung cancer. Oncogene. 2002; 21: 4129 -4136. [PubMed] .
- 39. Chen G, Bhojani MS, Heaford AC, Chang DC, Laxman B, Thomas DG, Griffin LB, Yu J, Coppola JM, Giordano TJ, Lin L, Adams D, Orringer MB, et al. Phosphorylated FADD induces NF-kappaB, perturbs cell cycle, and is associated with poor outcome in lung adenocarcinomas. Proc Natl Acad Sci U S A. 2005; 102: 12507 -12512. [PubMed] .
- 40. Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008; 8: 782 -798. [PubMed] .
- 41. Liu Y, Lang F, Xie X, Prabhu S, Xu J, Sampath D, Aldape K, Fuller G, Puduvalli VK. Efficacy of adenovirally expressed soluble TRAIL in human glioma organotypic slice culture and glioma xenografts. Cell Death Dis. 2011; 2: e121 [PubMed] .
- 42. Olsson M and Zhivotovsky B. Caspases and cancer. Cell Death Differ. 2011; 18: 1441 -1449. [PubMed] .
- 43. Gyrd-Hansen M and Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010; 10: 561 -574. [PubMed] .
- 44. Cheung HH, St Jean M, Beug ST, Lejmi-Mrad R, LaCasse E, Baird SD, Stojdl DF, Screaton RA, Korneluk RG. SMG1 and NIK regulate apoptosis induced by Smac mimetic compounds. Cell Death Dis. 2011; 2: e146 [PubMed] .
- 45. Hengartner MO. The biochemistry of apoptosis. Nature. 2000; 407: 770 -776. [PubMed] .
- 46. Ranger AM, Malynn BA, Korsmeyer SJ. Mouse models of cell death. Nat Genet. 2001; 28: 113 -118. [PubMed] .
- 47. Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009; 118: 5 -36. [PubMed] .
- 48. Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011; 2: e167 [PubMed] .
- 49. Ciavardelli D, Silvestri E, Viscovo AD, Bomba M, Gregorio DD, Moreno M, Ilio CD, Goglia F, Canzoniero LM, Sensi SL. Alterations of brain and cerebellar proteomes linked to Abeta and tau pathology in a female triple-transgenic murine model of Alzheimer's disease. Cell Death Dis. 2010; 1: e90 [PubMed] .
- 50. Rohn TT. The role of caspases in Alzheimer's disease; potential novel therapeutic opportunities. Apoptosis. 2010; 15: 1403 -1409. [PubMed] .
- 51. Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, et al. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999; 97: 395 -406. [PubMed] .
- 52. Shang YC, Chong ZZ, Wang S, Maiese K. Prevention of beta-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI 3-K/mTOR pathway, Bad, and Bcl-xL. Aging (Albany NY). 2012; 4: 187 -201. [PubMed] .
- 53. Noble W, Garwood C, Stephenson J, Kinsey AM, Hanger DP, Anderton BH. Minocycline reduces the development of abnormal tau species in models of Alzheimer's disease. FASEB J. 2009; 23: 739 -750. [PubMed] .
- 54. Banwait S, Galvan V, Zhang J, Gorostiza OF, Ataie M, Huang W, Crippen D, Koo EH, Bredesen DE. C-terminal cleavage of the amyloid-beta protein precursor at Asp664: a switch associated with Alzheimer's disease. J Alzheimers Dis. 2008; 13: 1 -16. [PubMed] .
- 55. Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009; 457: 981 -989. [PubMed] .
- 56. Rohn TT, Vyas V, Hernandez-Estrada T, Nichol KE, Christie LA, Head E. Lack of pathology in a triple transgenic mouse model of Alzheimer's disease after overexpression of the anti-apoptotic protein Bcl-2. J Neurosci. 2008; 28: 3051 -3059. [PubMed] .
- 57. Corona C, Pensalfini A, Frazzini V, Sensi SL. New therapeutic targets in Alzheimer's disease: brain deregulation of calcium and zinc. Cell Death Dis. 2011; 2: e176 [PubMed] .
- 58. Samantaray S, Knaryan VH, Guyton MK, Matzelle DD, Ray SK, Banik NL. The parkinsonian neurotoxin rotenone activates calpain and caspase-3 leading to motoneuron degeneration in spinal cord of Lewis rats. Neuroscience. 2007; 146: 741 -755. [PubMed] .
- 59. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998; 392: 605 -608. [PubMed] .
- 60. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004; 304: 1158 -1160. [PubMed] .
- 61. Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AY, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, Downward J, Mansfield L, Jat P, et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS One. 2008; 3: e2455 [PubMed] .
- 62. Simunovic F, Yi M, Wang Y, Macey L, Brown LT, Krichevsky AM, Andersen SL, Stephens RM, Benes FM, Sonntag KC. Gene expression profiling of substantia nigra dopamine neurons: further insights into Parkinson's disease pathology. Brain. 2009; 132: 1795 -1809. [PubMed] .
- 63. Barcia C, Ros CM, Annese V, Gomez A, Ros-Bernal F, Aguado-Yera D, Martinez-Pagan ME, de Pablos V, Fernandez-Villalba E, Herrero MT. IFN-gamma signaling, with the synergistic contribution of TNF-alpha, mediates cell specific microglial and astroglial activation in experimental models of Parkinson's disease. Cell Death Dis. 2011; 2: e142 [PubMed] .
- 64. Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H, Zeisler J, Squitieri F, Lin B, Bassett A, Almqvist E, et al. A worldwide study of the Huntington's disease mutation. The sensitivity and specificity of measuring CAG repeats. N Engl J Med. 1994; 330: 1401 -1406. [PubMed] .
- 65. Ross CA and Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004; 10 Suppl S10 -17. [PubMed] .
- 66. Bano D, Zanetti F, Mende Y, Nicotera P. Neurodegenerative processes in Huntington's disease. Cell Death Dis. 2011; 2: e228 [PubMed] .
- 67. Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996; 13: 442 -449. [PubMed] .
- 68. Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, Qin ZH, Aronin N, DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci U S A. 2001; 98: 12784 -12789. [PubMed] .
- 69. Graham RK, Deng Y, Carroll J, Vaid K, Cowan C, Pouladi MA, Metzler M, Bissada N, Wang L, Faull RL, Gray M, Yang XW, Raymond LA, et al. Cleavage at the 586 amino acid caspase-6 site in mutant huntingtin influences caspase-6 activation in vivo. J Neurosci. 2010; 30: 15019 -15029. [PubMed] .
- 70. Bhattacharyya NP, Banerjee M, Majumder P. Huntington's disease: roles of huntingtin-interacting protein 1 (HIP-1) and its molecular partner HIPPI in the regulation of apoptosis and transcription. FEBS J. 2008; 275: 4271 -4279. [PubMed] .
- 71. Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst CA, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V, Rasper DM, Roy S, Hayden MR, et al. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat Cell Biol. 2002; 4: 95 -105. [PubMed] .
- 72. Vucic S and Kiernan MC. Pathophysiology of neurodegeneration in familial amyotrophic lateral sclerosis. Curr Mol Med. 2009; 9: 255 -272. [PubMed] .
- 73. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX et al. Muta-tions in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993; 362: 59 -62. [PubMed] .
- 74. Pasinelli P and Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006; 7: 710 -723. [PubMed] .
- 75. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994; 264: 1772 -1775. [PubMed] .
- 76. Kim EK and Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010; 1802: 396 -405. [PubMed] .
- 77. Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron. 2002; 35: 1067 -1083. [PubMed] .
- 78. Bacman SR, Bradley WG, Moraes CT. Mitochondrial involvement in amyotrophic lateral sclerosis: trigger or target? Mol Neurobiol. 2006; 33: 113 -131. [PubMed] .
- 79. Meissner F, Molawi K, Zychlinsky A. Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc Natl Acad Sci U S A. 2010; 107: 13046 -13050. [PubMed] .
- 80. Pasinelli P, Houseweart MK, Brown RH Jr., Cleveland DW. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu, Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000; 97: 13901 -13906. [PubMed] .
- 81. Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng ZJ, Flegal K, O'Donnell C, Kittner S, Lloyd-Jones D, Goff DC Jr., Hong Y, et al. Heart disease and stroke statistics--2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006; 113: e85 -151. [PubMed] .
- 82. Downes CE and Crack PJ. Neural injury following stroke: are Toll-like receptors the link between the immune system and the CNS? Br J Pharmacol. 2010; 160: 1872 -1888. [PubMed] .
- 83. Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009; 14: 469 -477. [PubMed] .
- 84. Muralikrishna Adibhatla R and Hatcher JF. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic Biol Med. 2006; 40: 376 -387. [PubMed] .
- 85. Takadera T, Shiraishi Y, Ohyashiki T. Prostaglandin E2 induced caspase-dependent apoptosis possibly through activation of EP2 receptors in cultured hippocampal neurons. Neurochem Int. 2004; 45: 713 -719. [PubMed] .
- 86. Saleem S, Kim YT, Maruyama T, Narumiya S, Dore S. Reduced acute brain injury in PGE2 EP3 receptor-deficient mice after cerebral ischemia. J Neuroimmunol. 2009; 208: 87 -93. [PubMed] .
- 87. Ikeda-Matsuo Y, Tanji H, Narumiya S, Sasaki Y. Inhibition of prostaglandin E(2) EP3 receptors improves stroke injury via anti-inflammatory and anti-apoptotic mechanisms. J Neuroimmunol. 2011; .
- 88. Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, Krammer PH. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001; 8: 679 -686. [PubMed] .
- 89. Tian L, Rauvala H, Gahmberg CG. Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 2009; 30: 91 -99. [PubMed] .
- 90. Yu WR, Baptiste DC, Liu T, Odrobina E, Stanisz GJ, Fehlings MG. Molecular mechanisms of spinal cord dysfunction and cell death in the spinal hyperostotic mouse: implications for the pathophysiology of human cervical spondylotic myelopathy. Neurobiol Dis. 2009; 33: 149 -163. [PubMed] .
- 91. Cao J, Semenova MM, Solovyan VT, Han J, Coffey ET, Courtney MJ. Distinct requirements for p38alpha and c-Jun N-terminal kinase stress-activated protein kinases in different forms of apoptotic neuronal death. J Biol Chem. 2004; 279: 35903 -35913. [PubMed] .
- 92. Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci. 2004; 24: 7879 -7887. [PubMed] .
- 93. Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, Hyman BT, Yuan J, Kuida K, Flavell RA, Moskowitz MA. Caspase activation and neuroprotection in caspase-3- deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci U S A. 2002; 99: 15188 -15193. [PubMed] .
- 94. Schielke GP, Yang GY, Shivers BD, Betz AL. Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J Cereb Blood Flow Metab. 1998; 18: 180 -185. [PubMed] .
- 95. Betz AL, Yang GY, Davidson BL. Attenuation of stroke size in rats using an adenoviral vector to induce overexpression of interleukin-1 receptor antagonist in brain. J Cereb Blood Flow Metab. 1995; 15: 547 -551. [PubMed] .
- 96. Benchoua A, Guegan C, Couriaud C, Hosseini H, Sampaio N, Morin D, Onteniente B. Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci. 2001; 21: 7127 -7134. [PubMed] .
- 97. Pan J, Konstas AA, Bateman B, Ortolano GA, Pile-Spellman J. Reperfusion injury following cerebral ischemia: pathophysiology, MR imaging, and potential therapies. Neuroradiology. 2007; 49: 93 -102. [PubMed] .
- 98. Jung JE, Kim GS, Chen H, Maier CM, Narasimhan P, Song YS, Niizuma K, Katsu M, Okami N, Yoshioka H, Sakata H, Goeders CE, Chan PH. Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol. 2010; 41: 172 -179. [PubMed] .
- 99. Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010; 72: 19 -44. [PubMed] .
- 100. Cuadrado I, Fernandez-Velasco M, Bosca L, de Las Heras B. Labdane diterpenes protect against anoxia/reperfusion injury in cardiomyocytes: involvement of AKT activation. Cell Death Dis. 2011; 2: -e229. .
- 101. Gough DR and Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2011; 2: e213 [PubMed] .
- 102. Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003; 111: 1497 -1504. [PubMed] .
- 103. Nishida K and Otsu K. Cell death in heart failure. Circ J. 2008; 72 Suppl A A17 -21. [PubMed] .
- 104. Rice KC and Bayles KW. Molecular control of bacterial death and lysis. Microbiol Mol Biol Rev. 2008; 72: 85 -109. [PubMed] .
- 105. Häcker G, Kirschnek S, Fischer SF. Apoptosis in infectious disease: how bacteria interfere with the apoptotic apparatus. Med Microbiol Immunol. 2006; 195: 11 -19. [PubMed] .
- 106. Dao DN, Kremer L, Guerardel Y, Molano A, Jacobs WR Jr., Porcelli SA, Briken V. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infect Immun. 2004; 72: 2067 -2074. [PubMed] .
- 107. Tesh VL. Induction of apoptosis by Shiga toxins. Future Microbiol. 2010; 5: 431 -453. [PubMed] .
- 108. Carrero JA, Calderon B, Unanue ER. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J Immunol. 2004; 172: 4866 -4874. [PubMed] .
- 109. Ring A, Braun JS, Pohl J, Nizet V, Stremmel W, Shenep JL. Group B streptococcal beta-hemolysin induces mortality and liver injury in experimental sepsis. J Infect Dis. 2002; 185: 1745 -1753. [PubMed] .
- 110. Russell DG. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev. 2011; 240: 252 -268. [PubMed] .
- 111. Lee J, Hartman M, Kornfeld H. Macrophage apoptosis in tuberculosis. Yonsei Med J. 2009; 50: 1 -11. [PubMed] .
- 112. Lee J, Remold HG, Ieong MH, Kornfeld H. Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J Immunol. 2006; 176: 4267 -4274. [PubMed] .
- 113. Lopez M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol. 2003; 170: 2409 -2416. [PubMed] .
- 114. Driscoll JA, Brody SL, Kollef MH. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 2007; 67: 351 -368. [PubMed] .
- 115. Grassmé H, Jendrossek V, Gulbins E. Molecular mechanisms of bacteria induced apoptosis. Apoptosis. 2001; 6: 441 -445. [PubMed] .
- 116. Pan Y, Teng D, Burke AC, Haase EM, Scannapieco FA. Oral bacteria modulate invasion and induction of apoptosis in HEp-2 cells by Pseudomonas aeruginosa. Microb Pathog. 2009; 46: 73 -79. [PubMed] .
- 117. Burillo A and Bouza E. Chlamydophila pneumoniae. Infect Dis Clin North Am. 2010; 24: 61 -71. [PubMed] .
- 118. Dean D and Powers VC. Persistent Chlamydia trachomatis infections resist apoptotic stimuli. Infect Immun. 2001; 69: 2442 -2447. [PubMed] .
- 119. Fischer SF, Vier J, Kirschnek S, Klos A, Hess S, Ying S, Häcker G. Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. J Exp Med. 2004; 200: 905 -916. [PubMed] .
- 120. Gyles CL. Shiga toxin-producing Escherichia coli: an overview. J Anim Sci. 2007; 85: 13 Suppl E45 -62. [PubMed] .
- 121. Lingwood CA. Shiga toxin receptor glycolipid binding. Pathology and utility. Methods Mol Med. 2003; 73: 165 -186. [PubMed] .
- 122. Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SL, Magun BE. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol. 1997; 17: 3373 -3381. [PubMed] .
- 123. Johansson D, Kosovac E, Moharer J, Ljuslinder I, Brannstrom T, Johansson A, Behnam-Motlagh P. Expression of verotoxin-1 receptor Gb3 in breast cancer tissue and verotoxin-1 signal transduction to apoptosis. BMC Cancer. 2009; 9: 67 [PubMed] .
- 124. Blaser MJ and Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004; 113: 321 -333. [PubMed] .
- 125. Yamasaki E, Wada A, Kumatori A, Nakagawa I, Funao J, Nakayama M, Hisatsune J, Kimura M, Moss J, Hirayama T. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J Biol Chem. 2006; 281: 11250 -11259. [PubMed] .
- 126. Esen M, Schreiner B, Jendrossek V, Lang F, Fassbender K, Grassme H, Gulbins E. Mechanisms of Staphylococcus aureus induced apoptosis of human endothelial cells. Apoptosis. 2001; 6: 431 -439. [PubMed] .
- 127. Marriott HM, Ali F, Read RC, Mitchell TJ, Whyte MK, Dockrell DH. Nitric oxide levels regulate macrophage commitment to apoptosis or necrosis during pneumococcal infection. FASEB J. 2004; 18: 1126 -1128. [PubMed] .
- 128. Little SF and Ivins BE. Molecular pathogenesis of Bacillus anthracis infection. Microbes Infect. 1999; 1: 131 -139. [PubMed] .
- 129. Southwick FS and Purich DL. Intracellular pathogenesis of listeriosis. N Engl J Med. 1996; 334: 770 -776. [PubMed] .
- 130. Ulett GC and Adderson EE. Regulation of Apoptosis by Gram-Positive Bacteria: Mechanistic Diversity and Consequences for Immunity. Curr Immunol Rev. 2006; 2: 119 -141. [PubMed] .
- 131. Poxton IR, McCoubrey J, Blair G. The pathogenicity of Clostridium difficile. Clin Microbiol Infect. 2001; 7: 421 -427. [PubMed] .
- 132. He D, Hagen SJ, Pothoulakis C, Chen M, Medina ND, Warny M, LaMont JT. Clostridium difficile toxin A causes early damage to mitochondria in cultured cells. Gastroenterology. 2000; 119: 139 -150. [PubMed] .
- 133. Hay S and Kannourakis G. A time to kill: viral manipulation of the cell death program. J Gen Virol. 2002; 83: 1547 -1564. [PubMed] .
- 134. Danthi P. Enter the kill zone: initiation of death signaling during virus entry. Virology. 2011; 411: 316 -324. [PubMed] .
- 135. Brazeau E, Mahalingam R, Gilden D, Wellish M, Kaufer BB, Osterrieder N, Pugazhenthi S. Varicella-zoster virus-induced apoptosis in MeWo cells is accompanied by down-regulation of Bcl-2 expression. J Neurovirol. 2010; 16: 133 -140. [PubMed] .
- 136. Brune W. Inhibition of programmed cell death by cytomegaloviruses. Virus Res. 2011; 157: 144 -150. [PubMed] .
- 137. Pauleau AL, Larochette N, Giordanetto F, Scholz SR, Poncet D, Zamzami N, Goldmacher VS, Kroemer G. Structure-function analysis of the interaction between Bax and the cytomegalovirus-encoded protein vMIA. Oncogene. 2007; 26: 7067 -7080. [PubMed] .
- 138. Smith GB and Mocarski ES. Contribution of GADD45 family members to cell death suppression by cellular Bcl-xL and cytomegalovirus vMIA. J Virol. 2005; 79: 14923 -14932. [PubMed] .
- 139. Poncet D, Larochette N, Pauleau AL, Boya P, Jalil AA, Cartron PF, Vallette F, Schnebelen C, Bartle LM, Skaletskaya A, Boutolleau D, Martinou JC, Goldmacher VS, et al. An anti-apoptotic viral protein that recruits Bax to mitochondria. J Biol Chem. 2004; 279: 22605 -22614. [PubMed] .
- 140. Andoniou CE. Suicide watch: how cytomegalovirus interferes with the cell-death pathways of infected cells. Tissue Antigens. 2010; 76: 1 -8. [PubMed] .
- 141. Assrir N, Soussan P, Kremsdorf D, Rossignol JM. Role of the hepatitis B virus proteins in pro- and anti-apoptotic processes. Front Biosci. 2010; 15: 12 -24. [PubMed] .
- 142. Foo NC, Ahn BY, Ma X, Hyun W, Yen TS. Cellular vacuolization and apoptosis induced by hepatitis B virus large surface protein. Hepatology. 2002; 36: 1400 -1407. [PubMed] .
- 143. Liang X, Du J, Liu Y, Cui M, Ma C, Han L, Qu Z, Zhang Z, Sun Z, Zhang L, Chen YH, Sun W. The hepatitis B virus protein MHBs(t) sensitizes hepatoma cells to TRAIL-induced apoptosis through ERK2. Apoptosis. 2007; 12: 1827 -1836. [PubMed] .
- 144. Bouchard MJ and Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004; 78: 12725 -12734. [PubMed] .
- 145. Clippinger AJ, Gearhart TL, Bouchard MJ. Hepatitis B virus X protein modulates apoptosis in primary rat hepatocytes by regulating both NF-kappaB and the mitochondrial permeability transition pore. J Virol. 2009; 83: 4718 -4731. [PubMed] .
- 146. Lu YW, Tan TL, Chan V, Chen WN. The HBSP gene is expressed during HBV replication, and its coded BH3-containing spliced viral protein induces apoptosis in HepG2 cells. Biochem Biophys Res Commun. 2006; 351: 64 -70. [PubMed] .
- 147. Fischer R, Baumert T, Blum HE. Hepatitis C virus infection and apoptosis. World J Gastroenterol. 2007; 13: 4865 -4872. [PubMed] .
- 148. Ludwig S, Pleschka S, Planz O, Wolff T. Ringing the alarm bells: signalling and apoptosis in influenza virus infected cells. Cell Microbiol. 2006; 8: 375 -386. [PubMed] .
- 149. Holm GH and Gabuzda D. Distinct mechanisms of CD4+ and CD8+ T-cell activation and bystander apoptosis induced by human immunodeficiency virus type 1 virions. J Virol. 2005; 79: 6299 -6311. [PubMed] .
- 150. Cummins NW and Badley AD. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis. 2010; 1: e99 [PubMed] .
- 151. Nie Z, Bren GD, Vlahakis SR, Schimnich AA, Brenchley JM, Trushin SA, Warren S, Schnepple DJ, Kovacs CM, Loutfy MR, Douek DC, Badley AD. Human immunodeficiency virus type 1 protease cleaves procaspase 8 in vivo. J Virol. 2007; 816947 -6956. .
- 152. Nagata S. Apoptosis and autoimmune diseases. IUBMB Life. 2006; 58: 358 -362. [PubMed] .
- 153. Nagata S. Autoimmune diseases caused by defects in clearing dead cells and nuclei expelled from erythroid precursors. Immunological reviews. 2007; 220: 237 -250. [PubMed] .
- 154. Madkaikar M, Mhatre S, Gupta M, Ghosh K. Advances in autoimmune lymphoproliferative syndromes. Eur J Haematol. 2011; 87: 1 -9. [PubMed] .
- 155. Tischner D, Woess C, Ottina E, Villunger A. Bcl-2-regulated cell death signalling in the prevention of autoimmunity. Cell Death Dis. 2010; 1: e48 [PubMed] .
- 156. Fadeel B and Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005; 258: 479 -517. [PubMed] .
- 157. Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999; 286: 1735 -1738. [PubMed] .
- 158. Ren Y, Tang J, Mok MY, Chan AW, Wu A, Lau CS. Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum. 2003; 48: 2888 -2897. [PubMed] .
- 159. Munoz LE, Janko C, Schulze C, Schorn C, Sarter K, Schett G, Herrmann M. Autoimmunity and chronic inflammation - two clearance-related steps in the etiopathogenesis of SLE. Autoimmun Rev. 2010; 10: 38 -42. [PubMed] .
- 160. Bachmann HS, Otterbach F, Callies R, Nuckel H, Bau M, Schmid KW, Siffert W, Kimmig R. The AA genotype of the regulatory BCL2 promoter polymorphism ( 938C>A) is associated with a favorable outcome in lymph node negative invasive breast cancer patients. Clin Cancer Res. 2007; 13: 5790 -5797. [PubMed] .
- 161. Janes KA. RUNX1 and its understudied role in breast cancer. Cell Cycle. 2011; 10: 3461 -3465. [PubMed] .
- 162. Pujals A, Renouf B, Robert A, Chelouah S, Hollville E, Wiels J. Treatment with a BH3 mimetic overcomes the resistance of latency III EBV (+) cells to p53-mediated apoptosis. Cell Death Dis. 2011; 2: e184 [PubMed] .
- 163. Egea V, von Baumgarten L, Schichor C, Berninger B, Popp T, Neth P, Goldbrunner R, Kienast Y, Winkler F, Jochum M, Ries C. TNF-alpha respecifies human mesenchymal stem cells to a neural fate and promotes migration toward experimental glioma. Cell Death Differ. 2011; 18: 853 -863. [PubMed] .
- 164. Hervouet E, Vallette FM, Cartron PF. Impact of the DNA methyltransferases expression on the methylation status of apoptosis-associated genes in glioblastoma multiforme. Cell Death Dis. 2010; 1: e8 [PubMed] .
- 165. Stegh AH and DePinho RA. Beyond effector caspase inhibition: Bcl2L12 neutralizes p53 signaling in glioblastoma. Cell Cycle. 2011; 10: 33 -38. [PubMed] .
- 166. D'Errico I, Lo Sasso G, Salvatore L, Murzilli S, Martelli N, Cristofaro M, Latorre D, Villani G, Moschetta A. Bax is necessary for PGC1alpha pro-apoptotic effect in colorectal cancer cells. Cell Cycle. 2011; 10: 2937 -2945. [PubMed] .
- 167. D'Agostino L and Giordano A. A novel dual signaling axis for NSP 5a3a induced apoptosis in head and neck carcinoma. Oncotarget. 2011; 2: 1055 -1074. [PubMed] .
- 168. Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A, Gounon P, Allegra M, Bertolotto C, Bereder JM, Tartare-Deckert S, Bahadoran P, Auberger P, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011; 2: e199 [PubMed] .
- 169. Botton T, Puissant A, Cheli Y, Tomic T, Giuliano S, Fajas L, Deckert M, Ortonne JP, Bertolotto C, Tartare-Deckert S, Ballotti R, Rocchi S. Ciglitazone negatively regulates CXCL1 signaling through MITF to suppress melanoma growth. Cell Death Differ. 2011; 18: 109 -121. [PubMed] .
- 170. Jiang CC, Lai F, Tay KH, Croft A, Rizos H, Becker TM, Yang F, Liu H, Thorne RF, Hersey P, Zhang XD. Apoptosis of human melanoma cells induced by inhibition of B-RAFV600E involves preferential splicing of bimS. Cell Death Dis. 2010; 1: e69 [PubMed] .
- 171. Tamiya S, Etoh K, Suzushima H, Takatsuki K, Matsuoka M. Mutation of CD95 (Fas/Apo-1) gene in adult T-cell leukemia cells. Blood. 1998; 91: 3935 -3942. [PubMed] .
- 172. Dunkle A, Dzhagalov I, He YW. Cytokine-dependent and cytokine-independent roles for Mcl-1: genetic evidence for multiple mechanisms by which Mcl-1 promotes survival in primary T lymphocytes. Cell Death Dis. 2011; 2: e214 [PubMed] .
- 173. Gao N, Budhraja A, Cheng S, Liu EH, Chen J, Yang Z, Chen D, Zhang Z, Shi X. Phenethyl isothiocyanate exhibits antileukemic activity in vitro and in vivo by inactivation of Akt and activation of JNK pathways. Cell Death Dis. 2011; 2: e140 [PubMed] .
- 174. Heidari N, Hicks MA, Harada H. GX15-070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death Dis. 2010; 1: e76 [PubMed] .
- 175. Bleumink M, Kohler R, Giaisi M, Proksch P, Krammer PH, Li-Weber M. Rocaglamide breaks TRAIL resistance in HTLV-1-associated adult T-cell leukemia/lymphoma by translational suppression of c-FLIP expression. Cell Death Differ. 2011; 18: 362 -370. [PubMed] .
- 176. Campos-Sanchez E, Toboso-Navasa A, Romero-Camarero I, Barajas-Diego M, Sanchez-Garcia I, Cobaleda C. Acute lymphoblastic leukemia and developmental biology: A crucial interrelationship. Cell Cycle. 2011; 10: 3473 -3486. [PubMed] .
- 177. Boehrer S, Galluzzi L, Lainey E, Bouteloup C, Tailler M, Harper F, Pierron G, Ades L, Thepot S, Sebert M, Gardin C, de Botton S, Fenaux P, et al. Erlotinib antagonizes constitutive activation of SRC family kinases and mTOR in acute myeloid leukemia. Cell Cycle. 2011; 10: 3168 -3175. [PubMed] .
- 178. Greaves M. Leukemogenesis and ageing: 'fit for transformation'? Aging (Albany NY). 2011; 3: 79 -80. [PubMed] .
- 179. Crews L, Patrick C, Adame A, Rockenstein E, Masliah E. Modulation of aberrant CDK5 signaling rescues impaired neurogenesis in models of Alzheimer's disease. Cell Death Dis. 2011; 2: e120 [PubMed] .
- 180. Lee MH, Lin SR, Chang JY, Schultz L, Heath J, Hsu LJ, Kuo YM, Hong Q, Chiang MF, Gong CX, Sze CI, Chang NS. TGF-beta induces TIAF1 self-aggregation via type II receptor-independent signaling that leads to generation of amyloid beta plaques in Alzheimer's disease. Cell Death Dis. 2010; 1: e110 [PubMed] .
- 181. Cordeiro MF, Guo L, Coxon KM, Duggan J, Nizari S, Normando EM, Sensi SL, Sillito AM, Fitzke FW, Salt TE, Moss SE. Imaging multiple phases of neurodegeneration: a novel approach to assessing cell death in vivo. Cell Death Dis. 2010; 1: e3 [PubMed] .
- 182. Song B, Davis K, Liu XS, Lee HG, Smith M, Liu X. Inhibition of Polo-like kinase 1 reduces beta-amyloid-induced neuronal cell death in Alzheimer's disease. Aging (Albany NY). 2011; 3: 846 -851. [PubMed] .
- 183. Stanga S, Lanni C, Govoni S, Uberti D, D'Orazi G, Racchi M. Unfolded p53 in the pathogenesis of Alzheimer's disease: is HIPK2 the link? Aging (Albany NY). 2010; 2: 545 -554. [PubMed] .
- 184. Berry C, La Vecchia C, Nicotera P. Paraquat and Parkinson's disease. Cell Death Differ. 2010; 17: 1115 -1125. [PubMed] .
- 185. da Costa CA and Checler F. A novel parkin-mediated transcriptional function links p53 to familial Parkinson's disease. Cell Cycle. 2010; 9: 16 -17. [PubMed] .
- 186. Sassone J, Colciago C, Marchi P, Ascardi C, Alberti L, Di Pardo A, Zippel R, Sipione S, Silani V, Ciammola A. Mutant Huntingtin induces activation of the Bcl-2/adenovirus E1B 19-kDa interacting protein (BNip3). Cell Death Dis. 2010; 1: e7 [PubMed] .
- 187. Richards P, Didszun C, Campesan S, Simpson A, Horley B, Young KW, Glynn P, Cain K, Kyriacou CP, Giorgini F, Nicotera P. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington's disease. Cell Death Differ. 2011; 18: 191 -200. [PubMed] .
- 188. Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, Qin ZH, Aronin N, DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98: 12784 -12789. [PubMed] .
- 189. Genestine M, Caricati E, Fico A, Richelme S, Hassani H, Sunyach C, Lamballe F, Panzica GC, Pettmann B, Helmbacher F, Raoul C, Maina F, Dono R. Enhanced neuronal Met signalling levels in ALS mice delay disease onset. Cell Death Dis. 2011; 2: e130 [PubMed] .
- 190. Pasinelli P and Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006; 7: 710 -723. [PubMed] .
- 191. Meissner F, Molawi K, Zychlinsky A. Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107: 13046 -13050. [PubMed] .
- 192. Chauvier D, Renolleau S, Holifanjaniaina S, Ankri S, Bezault M, Schwendimann L, Rousset C, Casimir R, Hoebeke J, Smirnova M, Debret G, Trichet AP, Carlsson Y, et al. Targeting neonatal ischemic brain injury with a pentapeptide-based irreversible caspase inhibitor. Cell Death Dis. 2011; 2: e203 [PubMed] .
- 193. Lanzillotta A, Sarnico I, Ingrassia R, Boroni F, Branca C, Benarese M, Faraco G, Blasi F, Chiarugi A, Spano P, Pizzi M. The acetylation of RelA in Lys310 dictates the NF-kappaB-dependent response in post-ischemic injury. Cell Death Dis; 2010; 1: e96 .
- 194. Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, Krammer PH. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell death and differentiation. 2001; 8: 679 -686. [PubMed] .
- 195. Degli Esposti D, Sebagh M, Pham P, Reffas M, Pous C, Brenner C, Azoulay D, Lemoine A. Ischemic preconditioning induces autophagy and limits necrosis in human recipients of fatty liver grafts, decreasing the incidence of rejection episodes. Cell Death Dis. 2011; 2: e111 [PubMed] .
- 196. Machado NG, Alves MG, Carvalho RA, Oliveira PJ. Mitochondrial involvement in cardiac apoptosis during ischemia and reperfusion: can we close the box? Cardiovasc Toxicol. 2009; 9: 211 -227. [PubMed] .
- 197. Lopez-Neblina F, Toledo AH, Toledo-Pereyra LH. Molecular biology of apoptosis in ischemia and reperfusion. J Invest Surg. 2005; 18: 335 -350. [PubMed] .
- 198. Martins I, Galluzzi L, Kroemer G. Hormesis, cell death and aging. Aging (Albany NY). 2011; 3: 821 -828. [PubMed] .
- 199. Fujita T and Ishikawa Y. Apoptosis in heart failure. -The role of the beta-adrenergic receptor-mediated signaling pathway and p53-mediated signaling pathway in the apoptosis of cardiomyocytes. Circ J. 2011; 75: 1811 -1818. [PubMed] .
- 200. Kelk P, Abd H, Claesson R, Sandstrom G, Sjostedt A, Johansson A. Cellular and molecular response of human macrophages exposed to Aggregatibacter actinomycetemcomitans leukotoxin. Cell Death Dis. 2011; 2: e126 [PubMed] .
- 201. Gorvel L, Al Moussawi K, Ghigo E, Capo C, Mege JL, Desnues B. Tropheryma whipplei, the Whipple's disease bacillus, induces macrophage apoptosis through the extrinsic pathway. Cell Death Dis. 2010; 1: e34 [PubMed] .
- 202. Vazquez CL and Colombo MI. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ. 2010; 17: 421 -438. [PubMed] .
- 203. Hacker G, Kirschnek S, Fischer SF. Apoptosis in infectious disease: how bacteria interfere with the apoptotic apparatus. Medical microbiology and immunology. 2006; 195: 11 -19. [PubMed] .
- 204. Tesh VL. Induction of apoptosis by Shiga toxins. Future Microbiol. 2010; 5: 431 -453. [PubMed] .
- 205. Pan Y, Teng D, Burke AC, Haase EM, Scannapieco FA. Oral bacteria modulate invasion and induction of apoptosis in HEp-2 cells by Pseudomonas aeruginosa. Microb Pathog. 2009; 46: 73 -79. [PubMed] .
- 206. Calore F, Genisset C, Casellato A, Rossato M, Codolo G, Esposti MD, Scorrano L, de Bernard M. Endosome-mitochondria juxtaposition during apoptosis induced by H. pylori VacA. Cell Death Differ. 2010; 17: 1707 -1716. [PubMed] .
- 207. Halder UC, Bagchi P, Chattopadhyay S, Dutta D, Chawla-Sarkar M. Cell death regulation during influenza A virus infection by matrix (M1) protein: a model of viral control over the cellular survival pathway. Cell Death Dis. 2011; 2: e197 [PubMed] .
- 208. Krzyzowska M, Shestakov A, Eriksson K, Chiodi F. Role of Fas/FasL in regulation of inflammation in vaginal tissue during HSV-2 infection. Cell Death Dis. 2011; 2: e132 [PubMed] .
- 209. Knoll S, Furst K, Thomas S, Villanueva Baselga S, Stoll A, Schaefer S, Putzer BM. Dissection of cell context-dependent interactions between HBx and p53 family members in regulation of apoptosis: a role for HBV-induced HCC. Cell Cycle. 2011; 10: 3554 -3565. [PubMed] .
- 210. Brune W. Inhibition of programmed cell death by cytomegaloviruses. Virus Res. 2011; 157: 144 -150. [PubMed] .
- 211. Liang X, Du J, Liu Y, Cui M, Ma C, Han L, Qu Z, Zhang Z, Sun Z, Zhang L, Chen YH, Sun W. The hepatitis B virus protein MHBs(t) sensitizes hepatoma cells to TRAIL-induced apoptosis through ERK2. Apoptosis. 2007; 12: 1827 -1836. [PubMed] .
- 212. Fischer R, Baumert T, Blum HE. Hepatitis C virus infection and apoptosis. World journal of gastroenterology : WJG. 2007; 13: 4865 -4872. [PubMed] .
- 213. Abou-Kandil A, Chamias R, Huleihel M, Godbey WT, Aboud M. Role of caspase 9 in activation of HTLV-1 LTR expression by DNA damaging agents. Cell Cycle. 2011; 10: 3337 -3345. [PubMed] .
- 214. Mukerjee R, Claudio PP, Chang JR, Del Valle L, Sawaya BE. Transcriptional regulation of HIV-1 gene expression by p53. Cell Cycle. 2010; 9: 4569 -4578. [PubMed] .
- 215. Nagata S. Apoptosis and autoimmune diseases. Ann N Y Acad Sci. 2010; 1209: 10 -16. [PubMed] .
- 216. Qiao G, Li Z, Minto AW, Shia J, Yang L, Bao L, Tschopp J, Gao JX, Wang J, Quigg RJ, Zhang J. Altered thymic selection by overexpressing cellular FLICE inhibitory protein in T cells causes lupus-like syndrome in a BALB/c but not C57BL/6 strain. Cell Death Differ. 2010; 17: 522 -533. [PubMed] .
- 217. Eguchi K. Apoptosis in autoimmune diseases. Intern Med. 2001; 40: 275 -284. [PubMed] .
- 218. Kawakami A, Matsuoka N, Tsuboi M, Koji T, Urayama S, Sera N, Hida A, Usa T, Kimura H, Yokoyama N, Nakashima T, Ishikawa N, Ito K, et al. CD4+ T cell-mediated cytotoxicity toward thyrocytes: the importance of Fas/Fas ligand interaction inducing apoptosis of thyrocytes and the inhibitory effect of thyroid-stimulating hormone. Lab Invest. 2000; 80: 471 -484. [PubMed] .