




Introduction
Lamins A and C are intermediate filament proteins encoded by lamin A/C gene (LMNA) and constitute major components of nuclear lamina [1]. Mutations in LMNA have been shown to cause a wide range of human diseases collectively referred to as “laminopathies,”[2-6] from Hutchinson Gilford Progeria (premature aging syndrome), muscular dystrophy, to familial dilated cardiomyopathy (DCM). LMNA-related DCM is characterized by early onset of atrial fibrillation and conduction system disease, and subsequent progression to sudden cardiac death and heart failure [7, 8]. Indeed, LMNA mutations are the most common cause of familial DCM, accounting for 5-10% of overall familial DCM and up to 30-45% families with DCM and conduction system disease [9, 10]. Although the age of presentation in LMNA-related DCM can range from the first to sixth decade of life, almost all patients become symptomatic after age 60 [7, 11, 12]. Furthermore, LMNA-related DCM, especially in those associated with conductive system disease have a more malignant clinical course than other familial DCM due to a high rates of progressive heart failure and sudden cardiac death due to ventricular tachyarrhythmias [12-15]. Despite our increasing awareness on the importance of LMNA-relatedDCM, the mechanism of the disease as well as the therapeutic strategies to prevent the onset and progression of disease remain unclear.
Several animal models of LMNA mutations have been generated to provide initial insights into the pathophysiology for LMNA-related DCM[16-18]. In these animal models, either knock-in of mutant LMNA (dominant negative)[17] or knock-out of LMNA (haploinsufficieny) cause DCM in mice [16, 18], but variable cardiac phenotypes with or without conduction system disease were observed. The high mortality of the knockout mice also restricts the possibility of the whole animal study. As a result, the mechanisms by which different LMNA mutations cause DCM remain uncertain. An in-vitro platform of human cardiomyocytes derived from patients with different LMNA mutations would be extremely useful for understanding disease mechanism and for testing patient-specific therapies.
Latest breakthrough in the generation of human induced pluripotent stem cells (iPSC) from adult somatic tissues [19, 20] provides a unique opportunity to produce patient-specific cardiomyocytes for disease modeling and drug screening[21-23]. We and others [24, 25] have recently reported the use of human iPSC platform to model the disease phenotypes and mechanisms of Hutchinson-Gilford progeria syndrome, which is the most severe form of LMNA mutation that leads to premature aging and death. Here, we propose to recapitulate the disease phenotype, pathophysiology and drug screening for LMNA-related familial DCM in-vitro using cardiomyocytes derived from patient-specific human iPSCs (iPSC-CMs).
Results
Altered nuclear architecture and enhanced electric-stimulation induced apoptosis in LMNAR225X/WT dermal fibroblasts
We have established primary cultures of dermal fibroblasts from the proband (II-7) harboring the heterozygous R225X LMNA mutation and from a control individual (Figure 1). Compared with control dermal fibroblasts, LMNAR225X/WT dermal fibroblasts had reduced expression of the lamin A/C proteins (Figure 2A). Ultra-structural analysis using electronic microscopy revealed dramatic morphological alterations suggestive of nuclear senescence in LMNAR225X/WT dermal fibroblasts including focal loss of nuclear membrane, clustering of nuclear pores, large aggregates of highly condensed heterochromatin, bleb and micronucleus formation, and accumulation of mitochondria around the nuclear envelope (Figure 2B-I). Although these dysmorphic features of nuclear senescence, commonly seen in aging cells [26], could also be observed in the control fibroblasts with immunofluorescence analysis, they occurred in lower frequency (3.6 ± 3.6%, vs. 18.0 ± 1.8%, P<0.05) with less-severe morphological abnormalities in comparison with LMNAR225X/WT dermal fibroblasts (Figure 3A-E).
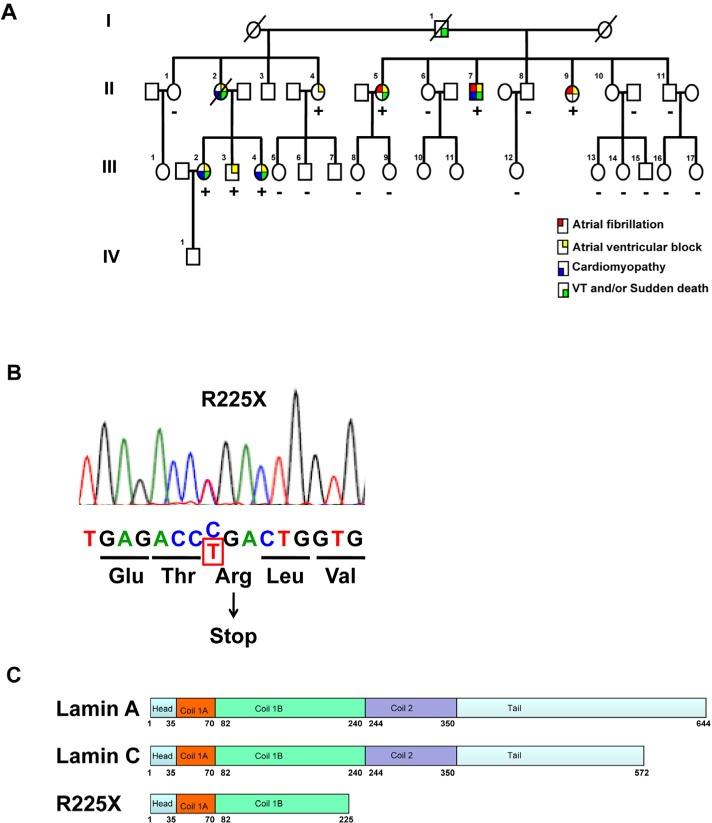
Figure 1. Clinical phenotype and LMNA genotype
(A) The pedigree of the proband (Patient II-7) with cardiac laminopathy featuring atrial fibrillation, atrioventricular block, dilated cardiomyopathy, ventricular tachyarrhythmia and sudden cardiac death. Four-generation family had 9 affected members showing autosomal-dominant cardiac disease. (See also table 1)(Square = male; circle = female; slash = deceased). A nonsense LMNA mutation (R225X) segregates with atrial fibrillation, atrioventricular block, dilated cardiomyopathy and ventricular tachyarrhythmia (“+” = LMNAR225X/WT, “-” = LMNAWT/WT). (B) The sequence chromatogram of the mutant allele of LMNA. The mutation altering amino acid sequence (in red) predicts a premature stop codon. (C) The schematic of lamin A/C proteins. Alternative splicing results at 572 and 664 amino acid lamin C and lamin A proteins. The R225X mutation results in a premature stop codon in the α-helical rod domain, thus a truncated lamin protein.
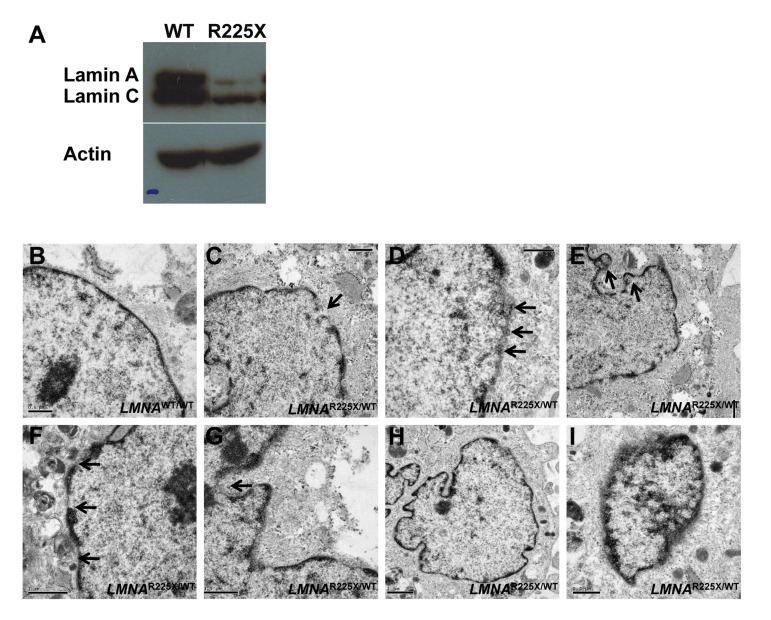
Figure 2. LMNAR225X/WT dermal fibroblasts showing nuclear defects and accelerated apoptosis upon electrical stimulation (A) The expressions of lamin A/C proteins in control and LMNAR225X/WT dermal fibroblasts with western blot analysis using anti-LMNA antibody targeting both N-terminus. (B-I) Electronic micrographs of nuclei of cultured dermal fibroblasts from controls and LMNAR225X/WT: (B) Normal nuclear envelope in control dermal fibroblast;(C) Focal loss of the nuclear membrane in LMNAR225X/WT dermal fibroblasts (arrow);(D) Clustering of nuclear pores;(E, F & G) Bleb and micronucleus formation; (H) Accumulation of mitochondria around the nuclear envelope; (H & I) Irregular shape nucleus, and condensed chromatin.
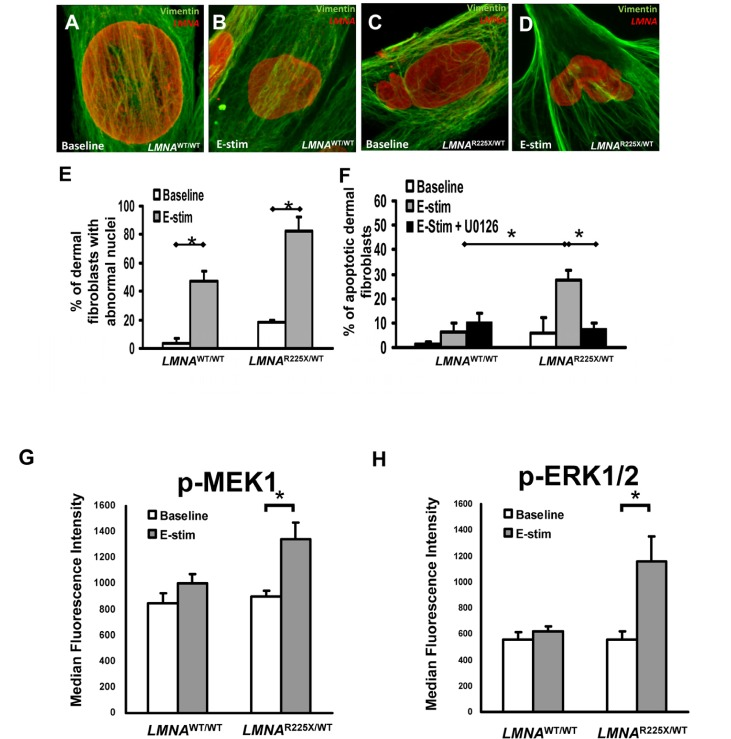
Figure 3. Effect of electrical stimulation on LMNAR225X/WT dermal fibroblasts (A-D) Immunofluorescence showing typical nuclear morphologies of control and LMNAR225X/WT dermal fibroblasts before and after electrical stimulation. (E) Effects of electrical stimulation on the percentage of control and LMNAR225X/WT dermal fibroblasts with nuclear blebs. (F) Effects of electrical stimulation and U0126 (ERK1/2 pathway blocker) on the apoptosis of control and LMNAR225X/WT dermal fibroblasts. The application of U0126, a highly selective blocker of the MEK1 pathway, nearly completely abolished the apoptotic effects of electrical stimulation on LMNAR225X/WT dermal fibroblasts. (G-H) Electrical stimulation-activates mitogen-activated protein kinases (MAPK) pathway in iPSC-CM: (G) MEK1 and (H) extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) in LMNAR225X/WT dermal fibroblasts, but not in control fibroblasts. *p- value <0.05.
Previous study in a lamin A-haploinsufficiency mouse model of DCM had suggested that reduction of lamin A/C proteins render premature apoptosis, particularly in electrically active cardiomyocytes [18.] Therefore, we applied electrical stimulation to cultured dermal fibroblasts to simulate an in-vivo cardiac electric field, and scored the fractions of abnormal nuclei present in control and LMNAR225X/WT dermal fibroblasts (Figure 3A-D). After electrical stimulation (at 6.6 V/cm, 1 Hz for 3 days), the percentage of cell with nuclear senescence (82 ± 9.7% vs. 47.4 ± 7.1%, P<0.05) and apoptosis (27.5 ± 4.1% vs. 6.5 ± 3.4%, P<0.05) were significantly higher in LMNAR225X/WT dermal fibroblasts as compared with control dermal fibroblasts (Figure 3E-F).
Mechanisms of electrical stimulation-induced apoptosis
We further explored the possible stress-related signaling pathway involved in electrical stimulation-induced apoptosis in LMNAR225X/WT dermal fibroblasts. Upon electrical stimulation, MEK1 and its downstream candidate, extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), were activated only in LMNAR225X/WT dermal fibroblasts, but not in control fibroblasts (Figure 3G-H). The application of U0126, a highly selective blocker of the MEK1-ERK1/2 pathway, nearly completely abolished the apoptotic effects of electrical stimulation on LMNAR225X/WT dermal fibroblasts (Figure 3F). These data suggest that increased nuclear senescence and apoptosis induced by electrical stimulation in LMNAR225X/WT dermal fibroblasts was mediated via the MEK1 pathway.
Generation of LMNAR225X/WT iPSC and cardiac differentiation
Despite the near-ubiquitous LMNA expression in most somatic cells, DCM is the most prominent phenotype of the R225X mutation. Therefore, we generated iPSCs from dermal fibroblasts isolated from the proband (II-7) to derive cardiomyocytes for recapitulating the cardiac phenotype of LMNA-related familial DCM in-vitro.
Altogether 4 clones of iPSCs were generated from the proband; however, chromosomal translocation was detected in 1 of these clones. As a result, the subsequent experiments were from the remaining 3 clones, which we did not detect any significant clone-to-clone variation. The genomic LMNA was sequenced in all iPSCs, and the expected mutation, R225X in LMNA was detected from the iPSCs generated from the dermal fibroblasts of the proband but not from those of the control. Besides, as we previously generated another iPSC line derived from DCM patient bearing frame-shift mutated LMNA [27], it was used as another LMNA halploinsuffiency model in the present study.
After cardiac differentiation, spontaneously beating outgrowths appeared approximately 14 to 21 days in embryoid bodies from both LMNAR225X/WT, LMNAFrameshift/WT and control iPSC lines (Figure 4A-C and Supplementary video I-III). The beating outgrowths were micro-surgically dissected out from the EBs and were dissociated into single cell clusters (Figure 4D-F, and Supplementary video IV-VI). Immunofluorescence analysis revealed the typical pattern of a cardiac-specific protein, α-actinin in beating cells from both LMNAR225X/WT, LMNAFrameshift/WT and control iPSC lines, confirming their cardiac identity (Figure 4G-I). Morphologically, there were no observable difference between the cardiomyocytes derived from the three iPSC lines, except the more prominent nuclear alterations of nuclei in LMNAR225X/WT andLMNAFrameshift/WT iPSC-CMs (Figure 4J-L).
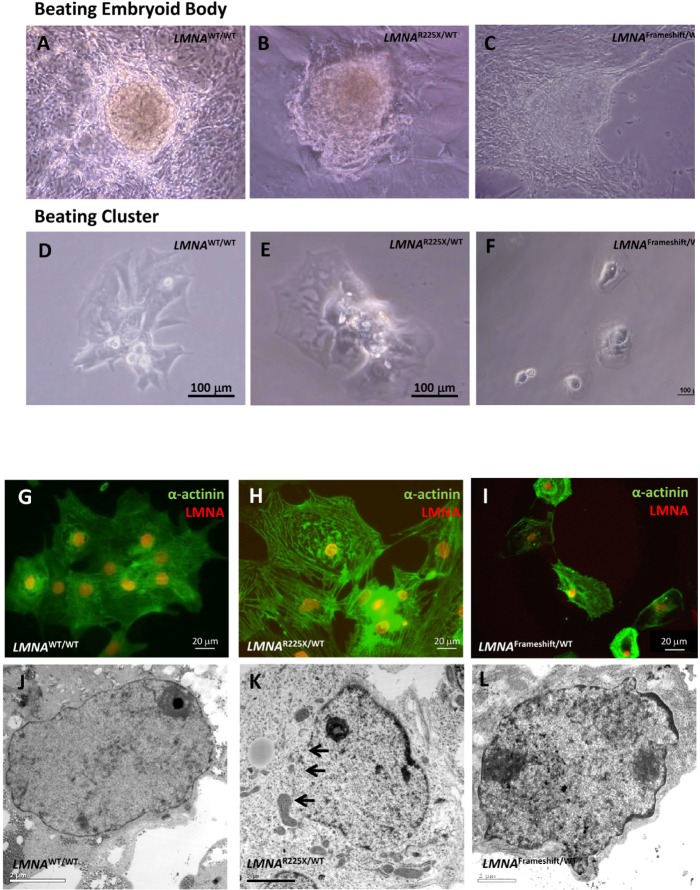
Figure 4. Cardiomyocytes derived from LMNAR225X/WT, LMNAFrameshift/WT and LMNAWT/WT iPSCs (A, B & C) Beating embryoid bodies derived from LMNAR225X/WT, LMNAFrameshift/WT and LMNAWT/WT iPSCs;(D, E & F) After micro-surgical dissection, spontaneously beating cell clusters were plated onto glass coverslips. Videos of these beating embryoid bodies and clusters were available in supplemental materials. (G, H & I) Immunofluorescence co-staining showing the expression of cardiac specific marker, α-actinin and lamin A/C in these cells, (J, K & L) Electronic microscopy of nucleus of cardiomyocytes derived from LMNAR225X/WT, LMNAFrameshift/WT and LMNAWT/WT iPSC line. The black arrows indicate the loss of nuclear envelope in the LMNAR225X/WT iPSC-derived cardiomyocytes.
Furthermore, we characterized the electrophysiological properties of LMNAR225X/WT iPSC-CMs using whole-cell patch clamp experiments. Figure 4I depicts representative single action potentials recorded from cardiomyocytes derived from LMNAR225X/WT and control iPSCs. According to action potential recording, cells were then classified into atrial- and ventricular-like cardiomyocytes. There were no significant differences in the proportion of atrial-like and ventricular-like cardiomyocytes between LMNAR225X/WT iPSCs and control. Furthermore, there were no significant differences in action potential duration at 50% and 90% repolarization (APD90) between the two groups (Data not shown).
Susceptibility of iPSC-CMs to electrical stimulation
Because electrical stimulation increased the fraction of cells with nuclear senescence as well as apoptosis in LMNAR225X/WT dermal fibroblasts, we further tested whether LMNAR225X/WT iPSC-CMs exhibited similar susceptibility to electrical stimulation. At baseline, there was no significant difference in the percentage of cardiomyocytes with nuclear senescence between the control and LMNAR225X/WT iPSC-CMs (3.1 ± 0.7 % vs. 6.8 ± 2.2%) (Figure 5A-B). Upon electrical stimulation, the percentage of nuclear senescence in LMNAR225X/WT and LMNAFrameshift/WT iPSC-CMs markedly increased to 48.2 ±4.0% (P<0.005 vs. baseline) and 52.3 ± 7.9% (P<0.05 vs. baseline) respectively and was significantly higher than those of the control iPSC-CMs (8.6±2.5%, P<0.01 in both mutated LMNA group; Figure 5B). More importantly, electrical stimulation resulted in a remarkably higher percentage of apoptotic cells in both LMNAR225X/WT and LMNAFramshift/WT iPSC-CMs compared with control (13.8±2.9% and 11.2±0.8% vs. 6.3±1.2%; n=3 P<0.05) as determined by APO-BrdU TUNEL assay quantitatively (Figure 6F and G). Co-staining of cardiac marker with TUNEL reaction further qualitatively verify the apoptotic events happened in LMNAR225X/WT and LMNAFramshift/WT iPSC-CMs upon electrical field stimulation.
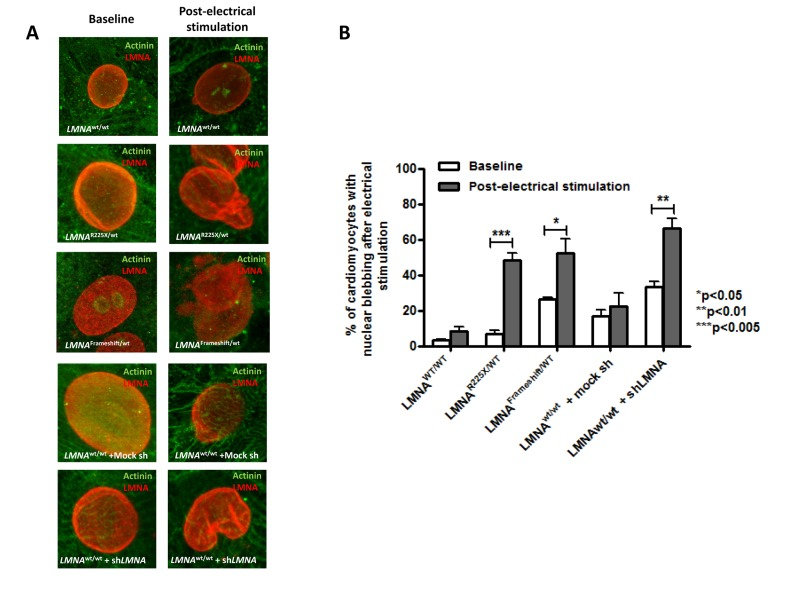
Figure 5. Electrical stimulation inducing nuclear abnormality in cardiomyocytes derived from LMNAR225X/WT &LMNAFrameshift/WT iPSCs (A) Representative immunofluorescence staining of cardiomyocytes derived from LMNAWT/WT iPSCs, LMNAR225X/WT iPSCs, LMNAFrameshift/WT iPSCs, LMNAWT/WT iPSCs treated with mock shRNA, and LMNAWT/WT iPSCs treated with shLMNA at baseline and after electrical stimulation. (B) The percentage of cardiomyocytes showed nuclear blebs.
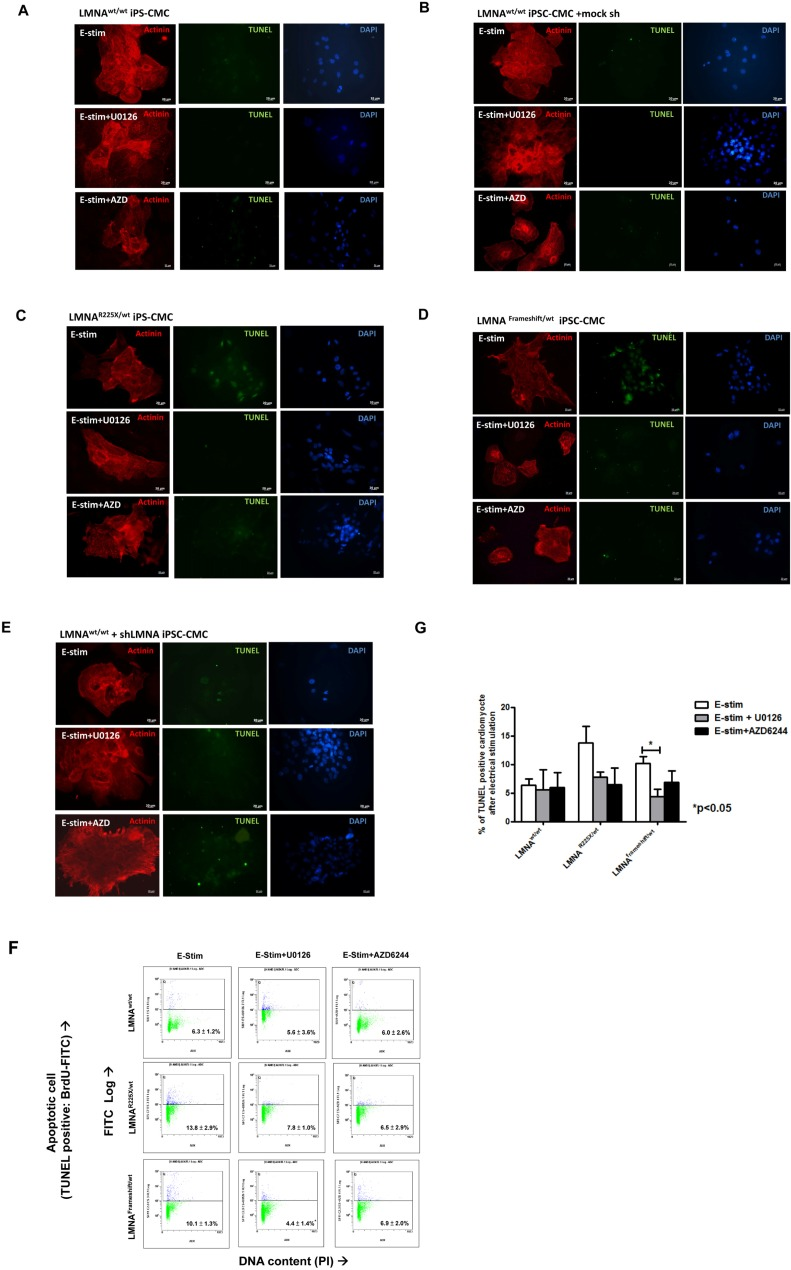
Figure 6. Electrical stimulation inducing apoptosis in cardiomyocytes derived from LMNAR225X/WT &LMNAFrameshift/WT iPSCs Representative TUNEL assay and co-immunofluorescence staining of alpha-actinin in cardiomyocytes derived from (A)LMNAWT/WT iPSCs. (B)LMNAR225X/WT iPSCs. (C)LMNAFrameshift/WT iPSCs. (D)LMNAWT/WT iPSCs treated with mock shRNA. (E)LMNAWT/WT iPSCs treated with shLMNA at baseline and after electrical stimulation. (F, G) Quantification of apoptotic cardiac differentiated iPSCs by APO-BrdU TUNEL assay. The percentage of cardiomyocytes with apoptosis was determined by FACS analysis by FL-1 positive gating. Unpaired t-test was performed between treatment and baseline n=3-5, *p-value<0.05.
To further verify whether lamin insufficiency contributes to the enhanced susceptibility of cardio-myocytes to electrical stimulation, we altered the expression of LMNA in control hiPSC-CMs by shRNA-mediated down-regulation (~60% as revealed by western blotting by probing of N-terminal LMNA A/C antibodies, data not shown). shRNA-treated control iPSC-CMs exhibited significantly higher percentage of nuclear senescence at baseline and after electrical stimulation (p<0.05) (Figure 5B). Likewise, shRNA mediated down-regulation of LMNA resulted in a significantly higher percentage of apoptotic cells than in control iPSC-CMs after electric stimulation (Figure 6E).
Intriguingly, the application of U0126 and selumetinib, AZD6244 (an anti-cancer drug in phase I-II clinical trial) to block MEK1 pathway in LMNAR225X/WT and LMNAFramshift/WT iPSC-CMs and shRNA treated control iPSC-CMs showed a trend that attenuated or even completely abolished the apoptotic effects of field electric stimulation on these lamin-deficient cardiomyocytes as seen in the dermal fibroblast (Figure 3 and 6) whereas 10 μM of U0126 significantly rescued the rate of apoptosis mediated by electrical stress (11.2 ± 0.8% vs. 4.4 ± 1.4%; n=3, P<0.05). On the other hand, although it has recently demonstrated that rapamycin reverses progeric phenotype in laminopathy, the addition of rapamycin (0.68 mM) did not alleviate the electrical stimulation mediated apoptosis of mutant LMNA iPSC-CMs (Supplementary Figure 3).
Discussion
LMNA-related DCM is the most common form of familial DCM encountered in clinical practice. Affected individuals are often asymptomatic at early stage except with typical ECG findings of low amplitude P waves, prolonged PR intervals, but relatively normal QRS complexes. As the disease progresses with age, patients develop atrial fibrillation, progressive conduction block and left ventricular dysfunction, and sudden cardiac death due to life-threatening ventricular tachyarrhythmias [7, 12, 15]. Since the first description of isolated DCM related to LMNA mutations by Fatkin et al in 1999[7], there are more than 40 LMNA mutations related to familial DCM have been reported. Despite of numerous clinical reports, the pathogenic mechanisms by which a defect in nuclear envelope link to the clinical DCM remain elusive.
In this study, we first demonstrated that primary dermal fibroblast from patients with nonsense LMNAR225X/WT mutation had reduced expression of the lamin A/C protein and exhibited prominent nuclear senescence as observed in aging cells. When LMNAR225X/WT dermal fibroblast exposed to external stimuli with electrical stimulation, markedly increased in the percentage of cells with nuclear senescence as well as apoptosis were observed as compared with control dermal fibroblast. These findings suggest that LMNAR225X/WT somatic cells have increased susceptibility to nuclear senescence and apoptosis after exposure to external physical stress. Furthermore, our pre-screening results showed that activation of stress response pathway with MEK1 contribute to increase apoptosis of LMNAR225X/WT dermal fibroblast after electrical stimulation.
Next, we generated disease-specific iPSC lines from the proband with LMNAR225X/WT and differentiated them together with another LMNA haploinsufficiency line, LMNAFrameshift/WT into cardiomyocytes. Interestingly, LMNAR225X/WT and LMNAFrameshift/WT iPSC-CMs showed normal phenotypes without significant nuclear abnormalities and electrophysiological properties at baseline as control iPSC-CMs. However, when these cardiomyocytes were subjected to electrical stimulation to mimic theirin-vivo host environment, they exhibited typical nuclear abnormalities resembling previous pathological findings from explanted human cardiomyocytes from individuals with LMNA-related DCM [28]. Furthermore, electrical stimulation significantly increased cellular apoptosis of LMNA haploinsufficiency iPSC-CMs, but not in control iPSC-CMs. The biological relevance of lamin A/C protein in cardiomyocytes was further verified by in-vitro knock-down of LMNA with adenoviral transient expression shRNA in control iPSC-CMs. Indeed, shRNA treated control iPSC-CMs mimicked the phenotypes changes as well as the susceptibility to apoptosis after electrical stimulation as LMNAR225X/WT and LMNAFrameshift/WT iPSC-CMs. More importantly, the increased susceptibility of apoptosis with electrical stimulation in the shRNA treated control- and the mutated LMNA iPSC-CMs could be attenuated or even completely abolished by pharmacological blockade of the MEK1/ERK1/2 pathway.
Currently, insights into the pathogenesis of LMNA-related DCM are mainly based on the observations from experimental animal models. In both lamin A/C-deficient (LMNA−/−)[16] and lamin A/C-insufficient (LMNA+/−)[18] mouse models, animals were born with normal functioning hearts, but eventually developed premature cardiac aging with various degree of atrioventricular block, atrial arrhythmias, DCM, and ventricular tachycardia, closely resembling to those observed in humans with heterozygous LMNA mutations. Although nuclear fragility secondary to lamin A/C-insufficiency has been postulated to render cardiomyocytes more susceptible to mechanical stress-induced damage and apoptosis [29], the application of mechanical stress to lamin A/C-insufficient (LMNA+/−) mice failed to enhance apoptosis or accelerate DCM.[30] On the contrary, histological studies of the hearts of both lamin A/C-insufficient (LMNA+/−), and lamin A/C-deficient (LMNA−/−) mice revealed typical nuclear abnormalities and apoptosis in cardiomyocytes. Interestingly, it appeared that electrically-active, conducting cardiomyocytes such as atrioventricular nodal cells exhibited more severe nuclear injuries and developed apoptosis much earlier than the non-conducting counterparts[18]. In concordance with these observations, previous clinical studies [7, 8] and our results showed that progressive conduction system disease is the early manifestation before the onset of DCM. In this study, family screening revealed that two of the affected subjects with LMNAR225X/WT mutation had subclinical conduction system disease before any clinical manifestation of DCM.
Accordingly, we hypothesized that lamin-insufficiency renders the cells more susceptible to apoptosis induced by electrical stimulation. Furthermore, we tested whether activation of stress response MEK1/ERK1/2 pathway contribute to the pathogenesis of apoptosis induced by electrical stimulation in LMNA-related DCM. Notably, ERK1/2, the downstream kinase of MEK1, is a member of the mitogen-activated protein kinases super family mediating cell proliferation and apoptosis depending on the stimuli and cell types. This is a particularly attractive explanation for LMNA-related DCM, as cardiomyocytes are constantly exposed to an alternating electrical field whereas cells in non-cardiac tissues might remain relatively unharmed, despite exhibiting the same nuclear abnormalities. Experimental studies using adult rat ventricular cardiomyocytes had demonstrated that under rapid electrical pacing, cultured cardiomyocytes had enhanced apoptosis as well as activation of ERK1/2 [31]. In a mouse model of another laminopathy, autosomal Emery-Dreifuss muscular dysfunction due to a missense H222P LMNA mutationalso manifests with DCM and atrioventricular conduction abnormality. In this model, ERK activation in heart muscle has been shown to play a key role in the pathogenesis of DCM [17]. More importantly, pharmacological inhibition of ERK activation prevented the development of DCM in these LMNAH222P/H222P mice [4]. In this study, pharmacology blockade of MEK1 pathway with U0126 and selumetinib significantly attenuated apoptosis in LMNAR225X/WT as well as LMNAFrameshift/WT iPSC-CMs after electrical stimulation, and selumetinib showed a consistent effect with a much greater efficacy (25 nM vs. 10 μM to rescue apoptosis). These findings in human iPSC-CM model provides further evidences to support the notion that pharmacological manipulation of MEK1 pathway might be a potential therapeutic target in LMNA-related DCM as reported in prior animal models [32]. Although selumetinib has been tested as anti-cancer therapy [33-35], its long-term safety and efficacy for treatment of LMNA-related DCM remain unclear.
Very recently, hyperactivation of the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway has been demonstrated in mouse models of laminopathies to be contributory to the cardiac and skeletal muscle defects [36, 37]. In fact, it has been shown that the pathway can determine the choice of cell fate at least in part between senescence and quiescence [38]. More importantly, inhibition of the pathway with rapamycin, a specific mTORC1 inhibitor alleviates cardiomyopathies in these mouse models [36, 37]. In a stark contrast, the administration of rapamycin to both LMNAR225X/WT and LMNAFrameshift/WT iPSC-CMs in our study failed to attenuate the electrical-stimulation induced apoptosis. This suggests that the observed attenuation of electrical-stimulation induced apoptosis of LMNA iPSC-CMs from ERK1/2 inhibition is not secondary to the indirect mTORC1 inhibition at least in these two LMNA mutations. In fact, it is well recognized that mutations in LMNA cause a protean range of human diseases with a high mutation-specific tissue-selectivity. Conceivably, different pathogenic mechanisms may be involved in different mutations. For instance, Hutchinson-Gilford progeria syndrome, the classical premature aging syndrome is due to a missense LMNA mutation leading to partially activates a cryptic splice donor site in exon 11, thereby producing an abnormal lamin A protein, progerin. The intracellular accumulation of progerin, as a toxin peptide, results in nuclear blebbing, mitotic abnormalities, replicative senescence, and accelerated telomere shortening [39]. In fact, the potential beneficial effects of rapamycin in human laminopathies were first demonstrated in fibroblasts from patients with Hutchinson-Gilford progeria syndrome [40]. Rapamycin promoting clearance of progerin by enhancing autophagy, has been demonstrated to abolish the development of nuclear bleeding and to delay the onset of cellular senescence of Hutchinson-Gilford progeria syndrome fibroblasts [40]. At a closer look, while the two LMNA mutations described in our study share very similar cardiac phenotypes (conduction abnormality, hear failure, ventricular tachyarrhythmia) with the previous reported LMNA mutation causing autosomal Emery-Dreifuss muscular dystrophy [41] presumably due to halo-insufficiency studied in the mouse models [17. 41, 42], none of our patients have clinical evidences of skeletal muscle abnormalities as in the original human laminopathy [41] and/or the derived mouse models of cardiac laminopathies [17. 41, 42]. Noteworthy, in the mouse model of the autosomal Emery-Dreifuss muscular dystrophy (missense H222P LMNA mutation), impaired authophagy due to the impaired mTORC1 pathway has been demonstrated which responded to mTORC1 inhibition [36]. Unlike the LMNAR225X mutation reported in our study, the H222P LMNA mutation in addition to halo-insufficiency may presumably produce and accumulate non-functioning lamin proteins similarly to progerin in Hutchinson Gilford progeria syndrome necessitating intrinsic autophagic mechanisms to clear them. However, the LMNAR225X mutation results in a premature stop-codon, which leads to the production of a very small truncated protein, thereby the predominant pathogenic mechanisms may be related to the loss of function of lamin due to halo-insufficiency rather than toxin peptide accumulation. Consequently, the enhanced autophagy due to the mTORC1 inhibition may have little beneficial effects to our cells. Given the diversity and high mutation-specific tissue-selectivity of LMNA related human diseases, human iPSCs appear to be a unique platform not only allowing investigating human diseases in a human platform in a mutation-specific manner, but also facilitating preclinical drug testing.
This study has several limitations. First, due to the lack of specific markers to allow effective sorting of different subset of cardiomyocytes such as nodal, atrial and ventricular myocytes, the effects of LMNA mutation as well as their in-vitro response to electrical stimulation remain unclear. Second, the results of this study may not be translated to LMNA-related DCM due to other mutations. Nevertheless, the ability to reproduce the disease phenotypes of LMNAR225X/WT iPSC-CMs using non-specific knock-down of LMNA with shRNA in control iPSC-CMs, suggesting that our results should be applicable to LMNA-related DCM due to haploinsufficiency.
In conclusion, we results show that patient-specific iPSC-CMs can be used to model the pathophysiology of LMNA-related DCM; and provide novel insight for potential therapeutic intervention for this most common form of familial DCM.
Methods
Clinical history and genetic phenotype
A 56 year-old Chinese man (II-7) presented to us with atrial fibrillation and cardioembolic stroke. He had history of progressive conduction system disease, evolving from 1st degree to 3rd degree atrioventricular block, and required permanent cardiac pacemaker implantation at age of 49 years (Table 1, and Figure 1A). Subsequently, he developed progressive heart failure and transthoracic echocardiogram revealed dilated right and left ventricles with impaired left ventricular ejection fraction of 35%. Then he underwent upgrading of his pacemaker to cardiac resynchronization therapy with defibrillator, and had received multiple device therapy for ventricular tachyarrhythmias during follow-up. Sequencing of the LMNA gene revealed a heterozygous single base exchange (672G→T) in exon 4, resulting in an R225X nonsense mutation (truncation in the coil 1B domain of both lamin A and C proteins), previously known to be associated with familial DCM (Figure 1B and IC)[43-45]. Indeed, he had a strong family history of DCM, conduction system disease and sudden death spanning 3 generations (Figure 1A). His father (I-1) suffered sudden death at age of 59, and one of his sisters (II-2) suffered from atrial fibrillation, heart failure and 3rd degree atrioventricular block with pacemaker implanted, who died suddenly and had documented ventricular fibrillation from her pacemaker memory. In addition, his two other sisters (II-5 and II-9) both had atrial fibrillation and 3rd degree atrioventricular block with permanent pacemaker implantation at 51 years and 48 years respectively. Subsequent screening of members of his family revealed asymptomatic 1st to 2nd degree atrioventricular block in 3 younger, apparently healthy family members (III-2, III-3, and III-4), in whom 24-hour ECG monitoring detected non-sustained ventricular tachyarrhythmia in two of them. Cardiac magnetic resonance imaging revealed patchy fibrosis over right ventricular myocardium (III-3 and III-4). Genetic testing showed that these family members had the same heterozygous LMNA mutation, confirming autosomal dominant inheritance in this family.
Table 1. Cardiac manifestations in affected subjects from a single extended family with LMNA R225X mutation
| Cardiac Manifestations (age of diagnosis in years) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Affected subjects | Sex | AV block | AF | VT/SCD | Cardiomyopathy | AICD/Pacemaker | Age of death | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I-1 | M | ? | ? | + (59) | ? | - | (59) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| II-2 | F | CHB (43) | + (46) | + (53) | + (43) | + (43) | (53) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| II-4 | F | 2° HB (53) | - | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| II-5 | F | CHB (51) | + (52) | - | - | + (51) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| II-7 | M | CHB (49) | + (49) | + (50) | + (51) | + (52) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| II-9 | F | CHB (48) | + (48) | - | - | + (48) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| III-2 | F | 2° HB, (43) | - | + (44) | - | + (44) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| III-3 | F | 1° HB, PR: 360ms (43) | - | - | - | - | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| III-4 | M | 1° HB, PR: 320ms (39) | - | + (39) | + (39) | - | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: 1° HB: first degree heart block; 2° HB: second degree heart block; AF: Atrial fibrillation; AICD: automatic implantable cardioverter defibrillator; AV block: atrio-ventricular block; CHB: complete heart block; PR: P-R interval; SCD: sudden cardiac death; VT: ventricular tachyarrhythmia. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Mitogen-Activated Protein Kinase (MAPK) phosphorylation analysis
Total cell lysate obtained from mutant and control primary fibroblast underwent electric stimulation from five individual experiment was subjected to MAPK pathway phosphorylation analysis using the Milliplex MAP 10plex MAPK/SAPK signaling kit (Millipore, St. Charles, Missouri, USA) according to manufacturer instructions.[46] Bio-Plex Manager software version 4.1.1 was used for data acquisition and analysis. Each lysate was measured in duplicated wells and the results were present as mean with standard error.
Generation of disease-specific induced pluripotent stem cells
For the generation of iPSCs, we recruited the proband (II-7) (LMNAR225X/WT) and another patient with frame-shift-mutated LMNA (contained a GCCA insertion at base 50 in lamin A/C gene, LMNA Frameshift/WT)along with one healthy age- and sex-matched control subject (LMNAWT/WT). The male patient (45 years old) bearing framshift-mutated LMNA was chosen also suffered from DCM and conduction system defectas previously described [27]. The study protocol of procurement of human tissue for the generation of iPSCs was approved by the local Institutional Review Board and was registered at the Clinical Trial Center, the University of Hong Kong (HKCTR-725, http://www.hkclinicaltrials.com).
After obtaining Informed consents from all participants, skin biopsies were taken, minced and then plated in 6-well culture dishes with 10% FBS medium as previously described[47]. Dermal fibroblasts growing out from the skin tissues were expanded and transduced with lentiviruses encoding OCT4, SOX2, KLF4, and c-MYC. Putative iPSC clusters were typically observed 14-21 days after lentiviral transduction, which were manually dissected and expanded in new Matrigel™-coated 6-well plates (Supplementary Figure 1). The authenticity of the human iPSCs was confirmed with the expression of a panel of pluripotent markers, transgene silencing, OCT4 promoter demethylation, and teratoma formation after inoculation into severe combined immunodeficiency mice (Supplementary Figure 1 and Result section in Supplementary Appendix). All the stem cell characterization of LMNAFrameshift/WT iPSC has been reported previously [27].
In vitro cardiac differentiation and characterization of cardiomyocytes
To induce cardiac differentiation, undifferentiated iPSCs were co-cultured with endoderm-like cells as described [48-51]. Briefly, undifferentiated iPSCs were dissociated into clumps using dispase (Invitrogen, CA, USA) and cultured in suspension using low attachment plates to form embryoid bodies. Embryoid bodies were transferred onto irradiated endoderm-like cells for co-culture. Beating outgrowths from iPSC embryoid bodies were micro-surgically dissected 20 days after induction of cardiac differentiation, followed by enzymatic dissociation into single isolated cardiomyocytes for further experiments. Rescued effect of electrical field-induced apoptosis of iPSC-CMs was studied by using mitogen-activated protein kinase kinase 1(MEK1)-inhibitor, 10 μM U0126 (dissolved in DMSO from Toricis), or 25 nM selumetinib, AZD6244 (dissolved in DMSO from Selleck Chemicals), an anti-cancer agent at phase I-II clinical trials; or 0.68 mM rapamycin (dissolved in DMSO from Sigma),[40] a specific inhibitor of the mTORC1 pathway. Standard immunofluorescence staining of alpha-actinin and electrophysiology analysis by patch clamping were performed to confirm their cardiac phenotypes [52-54].
shRNA mediated knockdown of LMNA
To verify the functional effect of LMNA in cardiomyocytes, the 18-nucleotide shRNA sequences designed to knockdown LMNA were tested in control iPSC-CM. shRNA against the common region of lamin A and C RNA position 609-627 from the start codon was cloned into pAd-GW/U6 vectors, and pAd-GFP was used as control. Adenovirus for shRNA against LMNA and control GFP vectors were produced using ViraPower Adenoviral Expression Kit (Invitrogen, CA, USA) and purified using Add-N-Pure Viral Purification kit (Applied Biological Materials Inc., Richmond, BC, Canada) according to manufacturer's instruction respectively. Viral titer was determined by AdEasy Viral titer kit (Stratagene, La Jolla, CA). A MOI of 10 was used for virus transduction. A knockdown efficiency of 30-40% of lamin A and C was observed.
Electrical field stimulation
To mimic the microenvironment of cardiomyocytes, iPSC-CMs were subjected to electrical stimulation. Cells were seeded on 13-mm glass cover slips (Nunc A/S, Rockilde, Denmark) and mounted inside a 6-well plate filled with corresponding medium. Electrical stimulation was then delivered to the cultured cell with carbon electrodes using an eight-channel C-Pace cell culture stimulator (Ion-Optics Co., Milton, MA, USA) at 6.6 V/cm, 1 Hz, 2 ms with alternating polarity for 4 hours [31]. Total number of cells was counted using standard cytometry by two blinded individuals before and after electrical stimulation.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay for apoptosis
TUNEL assay was performed using In Situ Cell Death Detection Kit, Fluorescein (Roche Applied Sciences, Mannheim, Germany) according to manufacturer's protocol. Cells were grown on glass coverslips, and after assigned treatment, cells were fixed and permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 minutes on ice. Cells were then incubated in TUNEL reaction mixture at 37°C for 60 minutes in a humidified dark chamber. Coverslips were mounted onto glycerol-based mountant. Images were acquired using Carl Zeiss fluorescence microscope with AxioVision 6.0 software (Zeiss GmbH, Gottingen, Germany).
Detection of apoptotic cells upon electrical stimulation was further quantified by the APO-BrdU TUNEL Assay Kit (Molecular Probes, Inc, Eugene, OR). The dissociated cells were fixed in 1% (w/v) paraformaldehyde on ice for 15 mins and then washed twice with DPBS at 300 × g for 5 mins. The fixed cells were stored in ice-cold 70% (v/v) ethanol at −20 ° C before nicked ends labeling reaction. Before incubation in TUNEL labeling solution for an hour at 37 ° C (10 μl reaction buffer, 0.75 μl TdT enzyme, 8 μl of BrdU and 31.25 μl dH2O), the cells were washed with wash buffer. After further washing by rinse buffer, the cells were stained with the Alexa Fluor® 488 dye-labeled anti-BrdU antibodies for half an hour. The propidium iodide/RNase A staining buffer was finally added to the cell prior to flow cytometry analysis. The positively labeled cells (green signal), a population with DNA fragmentation, were counted as % of apoptotic cells.
Statistical Analysis
Data are expressed as mean ± SEM. Statistic analysis was performed with the unpaired student t-test paired sample t-test as appropriate. Calculations were performed using SPSS software (version 12.0). A P-value < 0.05 was considered statistically significant.
Supplementary Materials
Acknowledgments
This work was supported by the Hong Kong Research Grant Council (HKU8/CRF/09 and T12-705/11 to Prof. Tse and Dr. Siu; and HKU 780110M to Prof. Tse) and the Singapore Agency for Technology, Science and Research (A Colman).
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Lin F and Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993; 268: 16321 -16326. [PubMed] .
- 2. Capell BC and Collins FS. Human laminopathies: nuclei gone genetically awry. Nature reviews. 2006; 7: 940 -952. .
- 3. Rankin J and Ellard S. The laminopathies: a clinical review. Clinical genetics. 2006; 70: 261 -274. [PubMed] .
- 4. Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009; 119: 1825 -1836. [PubMed] .
- 5. Burke B and Stewart CL. The laminopathies: the functional architecture of the nucleus and its contribution to disease. Annu Rev Genomics Hum Genet. 2006; 7: 369 -405. [PubMed] .
- 6. Dreesen O and Stewart CL. Accelerated aging syndromes, are they relevant to normal human aging? Aging (Albany NY). 2011; 3: 889 -895. [PubMed] .
- 7. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr., Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999; 341: 1715 -1724. [PubMed] .
- 8. Pan H, Richards AA, Zhu X, Joglar JA, Yin HL, Garg V. A novel mutation in LAMIN A/C is associated with isolated early-onset atrial fibrillation and progressive atrioventricular block followed by cardiomyopathy and sudden cardiac death. Heart Rhythm. 2009; 6: 707 -710. [PubMed] .
- 9. Fatkin D, Otway R, Richmond Z. Genetics of dilated cardiomyopathy. Heart Fail Clin. 2010; 6: 129 -140. [PubMed] .
- 10. Dellefave L and McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2010; 25: 198 -204. [PubMed] .
- 11. Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, Boucek MM, Lascor J, Moss AC, Li WL, Stetler GL, Muntoni F, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003; 41: 771 -780. [PubMed] .
- 12. Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, Mannarino S, Gambarin F, Favalli V, Grasso M, Agozzino M, Campana C, Gavazzi A, Febo O, Marini M, Landolina M, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008; 52: 1250 -1260. [PubMed] .
- 13. Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006; 354: 209 -210. [PubMed] .
- 14. Becane HM, Bonne G, Varnous S, Muchir A, Ortega V, Hammouda EH, Urtizberea JA, Lavergne T, Fardeau M, Eymard B, Weber S, Schwartz K, Duboc D. High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. Pacing Clin Electrophysiol. 2000; 23: 1661 -1666. [PubMed] .
- 15. van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbuchel H, de Visser M, Crijns HJ, Pinto YM. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med. 2005; 83: 79 -83. [PubMed] .
- 16. Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004; 113: 357 -369. [PubMed] .
- 17. Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest. 2007; 117: 1282 -1293. [PubMed] .
- 18. Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, Sherwood M, Branco DM, Wakimoto H, Fishman GI, See V, Stewart CL, Conner DA, Berul CI, Seidman CE, Seidman JG. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol. 2008; 44: 293 -303. [PubMed] .
- 19. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131: 861 -872. [PubMed] .
- 20. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007; 318: 1917 -1920. [PubMed] .
- 21. Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, Seyfarth M, Sinnecker D, Schomig A, Laugwitz KL. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2000; 363: 1397 -1409. [PubMed] .
- 22. Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011; 471: 225 -229. [PubMed] .
- 23. Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011; 471: 230 -234. [PubMed] .
- 24. Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, Stewart CL, Colman A. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011; 8: 31 -45. [PubMed] .
- 25. Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, Thompson J, Boue S, Fung HL, Sancho-Martinez I, Zhang K, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011; 472: 221 -225. [PubMed] .
- 26. Righolt CH, van 't Hoff ML, Vermolen BJ, Young IT, Raz V. Robust nuclear lamina-based cell classification of aging and senescent cells. Aging (Albany NY). 2011; 3: 1192 -1201. [PubMed] .
- 27. Ho JC, Zhou T, Lai WH, Huang Y, Chan YC, Li X, Wong NL, Li Y, Au KW, Guo D, Xu J, Siu CW, Pei D, Tse HF, Esteban MA. Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging (Albany NY). 2011; 3: 380 -390. [PubMed] .
- 28. Verga L, Concardi M, Pilotto A, Bellini O, Pasotti M, Repetto A, Tavazzi L, Arbustini E. Loss of lamin A/C expression revealed by immuno-electron microscopy in dilated cardiomyopathy with atrioventricular block caused by LMNA gene defects. Virchows Arch. 2003; 443: 664 -671. [PubMed] .
- 29. Hutchison CJ, Alvarez-Reyes M, Vaughan OA. Lamins in disease: why do ubiquitously expressed nuclear envelope proteins give rise to tissue-specific disease phenotypes? J Cell Sci. 2001; 114: 9 -19. [PubMed] .
- 30. Chandar S, Yeo LS, Leimena C, Tan JC, Xiao XH, Nikolova-Krstevski V, Yasuoka Y, Gardiner-Garden M, Wu J, Kesteven S, Karlsdotter L, Natarajan S, Carlton A, Rainer S, Feneley MP, Fatkin D. Effects of mechanical stress and carvedilol in lamin A/C-deficient dilated cardiomyopathy. Circ Res. 2010; 106: 573 -582. [PubMed] .
- 31. Kuramochi Y, Guo X, Sawyer DB, Lim CC. Rapid electrical stimulation induces early activation of kinase signal transduction pathways and apoptosis in adult rat ventricular myocytes. Exp Physiol. 2006; 91: 773 -780. [PubMed] .
- 32. Muchir A, Reilly SA, Wu W, Iwata S, Homma S, Bonne G, Worman HJ. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc Res. 2012; 93: 311 -319. [PubMed] .
- 33. Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S, Maloney L, Gordon G, Simmons H, Marlow A, Litwiler K, Brown S. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008; 26: 2139 -2146. [PubMed] .
- 34. Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, Marlow A, Hurley B, Lyssikatos J, Lee PA, Winkler JD, Koch K, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007; 13: 1576 -1583. [PubMed] .
- 35. Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy BA, Gilbert J, Mehra R, Moore DT, Sheikh A, Hoskins J, Hayward MC, Zhao N, O'Connor W, Weck KE, Cohen RB, et al. Phase II Efficacy and Pharmacogenomic Study of Selumetinib (AZD6244; ARRY-142886) in Iodine-131 Refractory Papillary Thyroid Carcinoma with or without Follicular Elements. Clin Cancer Res. 2012; 18: 2056 -2065. [PubMed] .
- 36. Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, Worman HJ. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. 2012; 4: 144ra102 .
- 37. Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, MacKay VL, An EH, Strong R, Ladiges WC, Rabinovitch PS, Kaeberlein M, Kennedy BK. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012; 4: 144ra103 .
- 38. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010; 2: 344 -352. [PubMed] .
- 39. Blagosklonny MV. Progeria, rapamycin and normal aging: recent breakthrough. Aging (Albany NY). 2011; 3: 685 -691. [PubMed] .
- 40. Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011; 3: 89ra58 .
- 41. Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E, Fromes Y, Toussaint M, Mura AM, Keller DI, Amthor H, Isnard R, Malissen M, Schwartz K, Bonne G. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005; 14: 155 -169. [PubMed] .
- 42. Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009; 18: 241 -247. [PubMed] .
- 43. Saga A, Karibe A, Otomo J, Iwabuchi K, Takahashi T, Kanno H, Kikuchi J, Keitoku M, Shinozaki T, Shimokawa H. Lamin A/C gene mutations in familial cardiomyopathy with advanced atrioventricular block and arrhythmia. Tohoku J Exp Med. 2009; 218: 309 -316. [PubMed] .
- 44. van Tintelen JP, Hofstra RM, Katerberg H, Rossenbacker T, Wiesfeld AC, du Marchie Sarvaas GJ, Wilde AA, van Langen IM, Nannenberg EA, van der Kooi AJ, Kraak M, van Gelder IC, van Veldhuisen DJ, Vos Y, van den Berg MP. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am Heart J. 2007; 154: 1130 -1139. [PubMed] .
- 45. Jakobs PM, Hanson EL, Crispell KA, Toy W, Keegan H, Schilling K, Icenogle TB, Litt M, Hershberger RE. Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J Card Fail. 2001; 7: 249 -256. [PubMed] .
- 46. Lee YK, Ng KM, Lai WH, Man C, Lieu DK, Lau CP, Tse HF, Siu CW. Ouabain facilitates cardiac differentiation of mouse embryonic stem cells through ERK1/2 pathway. Acta Pharmacol Sin. 2011; 32: 52 -61. [PubMed] .
- 47. Lai WH, Ho JC, Lee YK, Ng KM, Au KW, Chan YC, Lau CP, Tse HF, Siu CW. ROCK inhibition facilitates the generation of human-induced pluripotent stem cells in a defined, feeder-, and serum-free system. Cell Reprogram. 2010; 12: 641 -653. [PubMed] .
- 48. Mummery CL, Ward D, Passier R. Differentiation of human embryonic stem cells to cardiomyocytes by coculture with endoderm in serum-free medium. Current protocols in stem cell biology. 2007; Chapter 1:Unit 1F 2. .
- 49. Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, van der Heyden M, Opthof T, Pera M, de la Riviere AB, Passier R, Tertoolen L. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003; 07: 2733 -2740. [PubMed] .
- 50. Lee YK, Ng KM, Lai WH, Chan YC, Lau YM, Lian Q, Tse HF, Siu CW. Calcium homeostasis in human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Rev. 2011; 7: 976 -986. [PubMed] .
- 51. Ng KM, Lee YK, Lai WH, Chan YC, Fung ML, Tse HF, Siu CW. Exogenous expression of human apoA-I enhances cardiac differentiation of pluripotent stem cells. PLoS One. 2011; 6: e19787 [PubMed] .
- 52. Lee YK, Ng KM, Chan YC, Lai WH, Au KW, Ho CY, Wong LY, Lau CP, Tse HF, Siu CW. Triiodothyronine promotes cardiac differentiation and maturation of embryonic stem cells via the classical genomic pathway. Mol Endocrinol. 2010; 24: 1728 -1736. [PubMed] .
- 53. Ng KM, Lee YK, Chan YC, Lai WH, Fung ML, Li RA, Siu CW, Tse HF. Exogenous expression of HIF-1 alpha promotes cardiac differentiation of embryonic stem cells. J Mol Cell Cardiol. 2010; 48: 1129 -1137. [PubMed] .
- 54. Au KW, Liao SY, Lee YK, Lai WH, Ng KM, Chan YC, Yip MC, Ho CY, Wu EX, Li RA, Siu CW, Tse HF. Effects of iron oxide nanoparticles on cardiac differentiation of embryonic stem cells. Biochem Biophys Res Commun. 2009; 379: 898 -903. [PubMed] .