




Introduction
Senescence is a biological state in which the cells have an irreversible proliferative arrest while they continue to be metabolic active. The senescent phenotype is acquired in vitro after multiple rounds of cell division (replicative senescence) or upon oncogenes activation or oxidative stress (stress-induced premature senescence) [1-8]. The mechanisms underlying replicative senescence include telomere shortening, permanent growth arrest from G1 to S phase of the cell cycle, enhanced beta-galactosidase activity and increased expression of key mediators including p53, promyelocitic leukemia protein (PML), p16INK4a and p19Arf [1,9-12]. While cellular senescence is considered a protective mechanism against cancer, it has also been hypothesized that the progressive accumulation of senescent cells in some tissues may contribute to several age-related diseases and organismal aging [13-15]. Progressive DNA damage and mitochondrial decline are both considered to be prime instigators of natural ageing. These two pathways have been viewed largely in isolation. However, recent studies have revealed a molecular circuit that directly links DNA damage to compromised mitochondrial biogenesis and function via p53 [16-22]. Furthermore, the mitochondrial function decline is considered a major mechanism underlying senescence. Damaged mitochondria not only produce less ATP but also generate increased amounts of reactive oxygen species and display a greater propensity to trigger apoptosis [23-29].
The aging process, especially of the skin, is governed by changes in the epidermal, dermo-epidermal and dermal compartments. Skin function is mediated primarily by the structure of the epidermal and dermal layers [30,31]. The highly cellular, yet avascular, epidermis forms a barrier, which prevents both water and heat loss and the entry of pathogenic organisms. In contrast, the dermis is both vascularised and relatively acellular. The two layers are joined by a compositionally complex undulating dermo-epidermal junction (DEJ) in which basal epidermal keratinocytes are secured to a type IV collagen-rich basement membrane by hemidesmosomes and the dermis is anchored by collagen VII fibrils and fibrillin-rich microfibril bundles [32-36]. Sparsely distributed, dermal fibroblasts are thought to be responsible for synthesizing the three major groups of dermal extracellular matrix (ECM) proteins and collectively these ECM assemblies not only dominate the structure and function of the dermis, but via aberrant remodeling, mediate the changing function of aging skin. Aged skin is characterized by a flattening of the dermo-epidermal junction, a marked atrophy and a loss of elasticity of the dermal connective tissue [37], associated with a reduction and disorganization of its major extracellular matrix components, such as collagen and other elastic fibers [38], proteoglycans and glycosaminoglycans [39]. Some studies proposed that the consequences of ECM changes affected fibroblast phenotypic behavior in the dermis asserting that this cell situated in a damaged matrix were no longer subjected to mechanical stretching via integrin-collagen interactions [40-42]. Alteration of fibroblast properties, associated with aging or senescence, have been studied either in long-term cell cultures [43-48], or by using fibroblasts from skin biopsies derived from donors with increasing age [49]. The observed age related changes in fibroblast include cell morphology, metabolism [50], decline in the production of extracellular matrix proteins such as type I and III collagens [41], and overexpression of proteases involved in the degradation of the extracellular matrix [51]. All senescing cells undergo profound changes in gene expression. Altered gene expression gives rise to the senescent phenotype, and is well established as part of the mechanisms and pathways that activate the senescence program in cells. However, the factors responsible for the alterations of gene expression during senescence remain elusive.
MicroRNAs (miRNAs) are key modulators of gene expression in various biological and pathological processes. These RNAs, ~22 nucleotides in size, act as sequence guides that direct Argonaute protein complexes to mRNAs, where they decrease protein synthesis through translational repression or mRNA degradation and thereby influence many basic cellular processes and diseases [52-58]. Changes in miRNA expression levels occur in cellular senescence and organismal aging [59-64], and have been linked to changes in levels of mRNAs that are putative targets of specific miRNAs [65-67]. The regulation of miRNAs expression during senescence and aging represents an emerging and the list of miRNAs and corresponding target mRNAs involved in these pathways is rapidly increasing [68-73]. Recently, in a model of in vitro replicative senescence of normal human epidermal keratinocytes neonatal (HEKn), we have identified miR-191 as an anti-proliferative and senescence-associated miRNA [74] and also a ΔNp63α-miRNAs regulatory loop that represents a “stemness master gene”-mediated strategy to promote proliferation and to counteract senescence [75]. Several studies have identified specific sets of miRNAs up-regulated in keratinocytes and fibroblasts replicative senescence [75-78], among these we focused on miR-152 and miR-181a. MiR-152, although has been detected in replicative-induced fibroblast senescence screenings [76], has never been studied to identify its targets and its role in the senescence process. MiR-181a expression is up-regulated during keratinocytes replicative senescence and its overexpression is sufficient per se to induce cellular senescence [75].
Here, we have investigated the role of miR-152 and miR-181a in replicative senescence of primary human dermal fibroblasts (HDFn) providing evidence that the expression of these miRNAs in young proliferating fibroblasts is sufficient to induce senescence markers. We have also identified as new targets involved in this process, itga5 and col16a1, whose decrease is probably involved in ECM aberrant remodelling of aged skin.
Results
Induction of replicative senescence in human dermal fibroblasts
To generate a model of replicative senescence in which to assess miRNAs expression profile, proliferating primary HDFn cells were monitored through serial passaging. The proliferating or senescent state of the cells was assessed morphologically. A growth curve that calculated the population doublings at each passage was generated (Figure 1A). After sixteen passages (p), the proliferation rate slows down, leading to a complete block after seventeen passages. As shown in figure 1B, at p16 senescent cells stained positive for SA-β-galactoside compared to the proliferating population at p1. Between p1 and p16 we also observed a significant decrease in the percent of BrdU incorporating cells (from 35% to 8%, Figure 1C) indicating cell cycle G1-arrest. Finally, as further control, a western blot analysis was performed on protein extracts from HDFn cells collected at p1 and p16 passages. As expected, p53 and p16INK4a, which are senescence markers, are induced during passages (Figure 1D). In a previous study Yong Wang and colleagues showed, using microarray approach, that miR-152 is among the miRNAs more upregulated in replicative-induced fibroblast senescence [76] but it has never been studied to identify its targets and its role in the senescence process. In addition, by in silico analyses we found that miR-152 and miR-181a, the latter involved in keratinocytes-induced senescence [75], have as putative targets many mRNA involved in cell adhesion and extracellular matrix remodelling, therefore we decide to study the role of these miRNAs in our experimental system. MiRNAs expression levels were assessed and compared, by Real Time-qPCR, in RNA samples purified from proliferating (p1) and senescent (p16) HDFn. We confirmed, by real-time PCR that both, miR-152 and miR-181a, are significantly upregulated during fibroblasts replicative-induced senescence (Figure 1E).
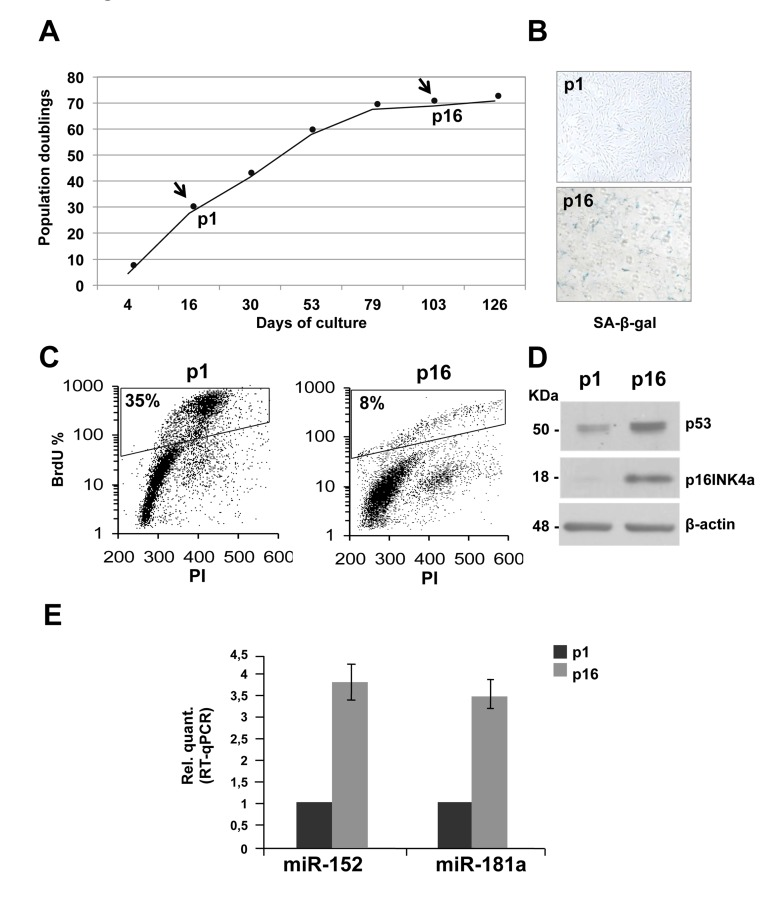
Figure 1. Replicative senescence in human dermal fibroblasts (HDFn) (A) Growth curve of cultured HDFn. (B) SA-β-gal staining of HDFn cells at p1 and p16 in culture. (C) HDFn cultured cells at p1 and p16 (p, passage) were subjected to a 4h BrdU-pulse, then collected, PI stained and analyzed by flow cytometry. BrdU positive cells are indicated as S phase fluorescent population and are assessed by PI staining of DNA content of 2n or 4n (fixed to values of 200 and 400 in the plots). (D) Western blots performed on proteins extracts from HDFn at p1 and p16 showing the analysis of some senescence markers, such as p53 and p16 levels. β-actin was used as loading control. (E) Real Time RT-qPCR was employed to analyze the expression levels of miR-152 and miR-181a in HDFn cells at a growing number of passages in culture (p1-p16). Values reported are the average ± SD of three independent experiments.
miR-152 and miR-181a induce senescence in human dermal fibroblasts
To investigate the role of miR-152 and miR-181a in human dermal fibroblast, we set up overexpression by transfection with pre-miR-152, pre-miR-181a, or scrambled control sequence in proliferating HDFn cells. The miRNAs were overexpressed 3-5 folds to mimic their physiological expression level during senescence. Ninety-six hours after transfection we observed a decrease about 40% in BrdU incorporation in the presence of miR-152 and miR-181a (respectively 18% and 19% versus 26% of scrambled control, Figure 1A) indicating a cell cycle G1-arrest. Overexpression of miR-152 and miR-181a had additional effects in HDFn cells, including the induction of the senescence markers p53 and p16INK4a(Figure 2B) and an increase in the number of cells positive for SA-β-galactosidase staining as shown by the images and blue cell quantification in Figure 2C and 2D. These results suggest that miR-152 and -181a overexpression, is sufficient per se to induce cellular senescence.
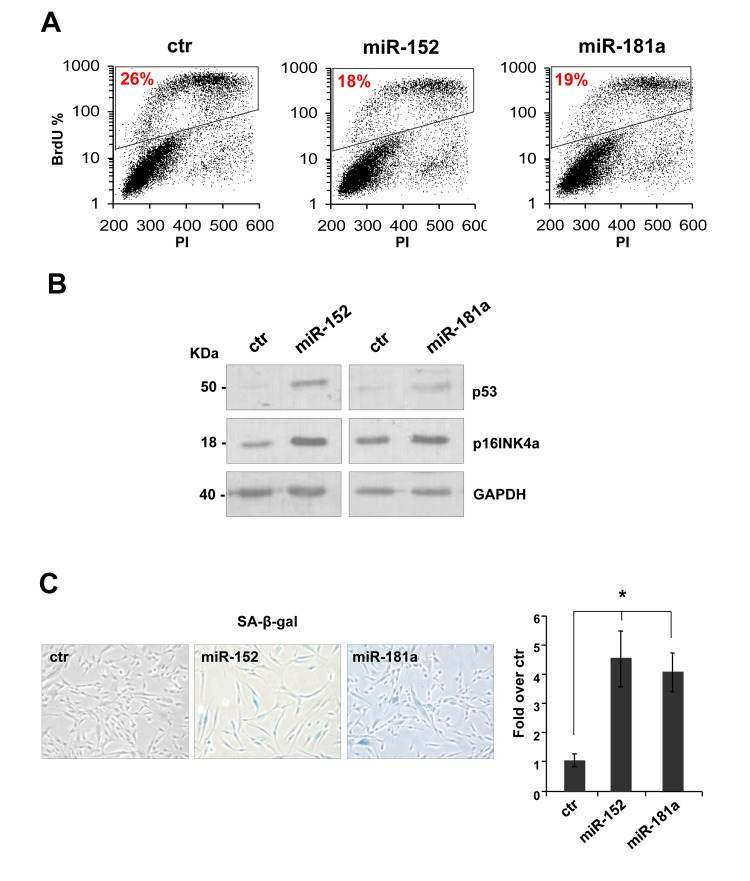
Figure 2. miR-152 and miR-181a induce cellular senescence in HDFn cells (A) 96 h after transfection of HDFn cells with scramble control (Ctr), miR-152 or miR-181a sequence, cells were subjected to a 4h BrdU-pulse, then collected, PI stained and analyzed by flow cytometry as described in Figure 1. (B) Western blot analysis of protein extract of HDFn transfected with miR-152 and miR-181a versus scramble control sequence (ctr). MiR-152and miR-181a overexpression increase p53 and p16INK4 protein level. β-actin was used as loading control. (C-D) SA-β-gal staining and quantification by blue cells counting/field (as fold over control). Values reported are average ± SD of three independent stains. *p-Value <0.01 by Student's t test.
miR-152 represses ITGA5 expression and controls HDFn cell adhesion
To investigate the role of miR-152 on ECM remodeling, we started a detailed analysis of its putative molecular targets. From an in silico prediction, by TargetScan 6.2 database, we selected ITGA5-3'UTR as putative target of miR-152. These 3'UTR harbors at least one miR-152 target site that is highly conserved among vertebrates (Figure 3A). As shown in figure 3B, real time-qPCR analysis showed a significant decrease (about 30%) of ITGA5 mRNA level between p1 and p16 fibroblasts. The down-regulation of miR-152 putative target, was also possible confirmed at the protein level, in fact a western blot analysis showed a decrease of itga5 protein level in HDFn cells from p1 to p16 (Figure 3C). The observed inverse correlation between miR-152 expression and its putative target strengthened the hypothesis of its direct regulation. Integrins consist of 18 α- and 8 β-glycoprotein subunits, which form 24 distinct heterodimeric transmembrane receptors. These receptors bind to ECM proteins such as fibronectin to transport signals bidirectional across the cell membrane, allowing cells to respond to environmental changes [79,80]. Furthermore, integrin binding to ECM is not only required for transducing signals from the matrix to cells, but this interaction also initiates responses that allow the cells to organize and remodel the matrix [81], this features is very limited in aged tissues and in particular in aged skin. The mesenchymal integrins α5 (ITGA5) and β1 form heterodimers to mediate cell adhesion to fibronectin [82]. Knock-out of ITGA5 in mice results in embryonic lethality [83]. In human epatocarcinoma cells, ITGA5 promotes cell adhesion and migration on fibronectin through activating focal adhesion kinase [84]. This findings indicate that ITGA5 expression plays an important role to enhance cell adhesion to and migration on fibronectin.
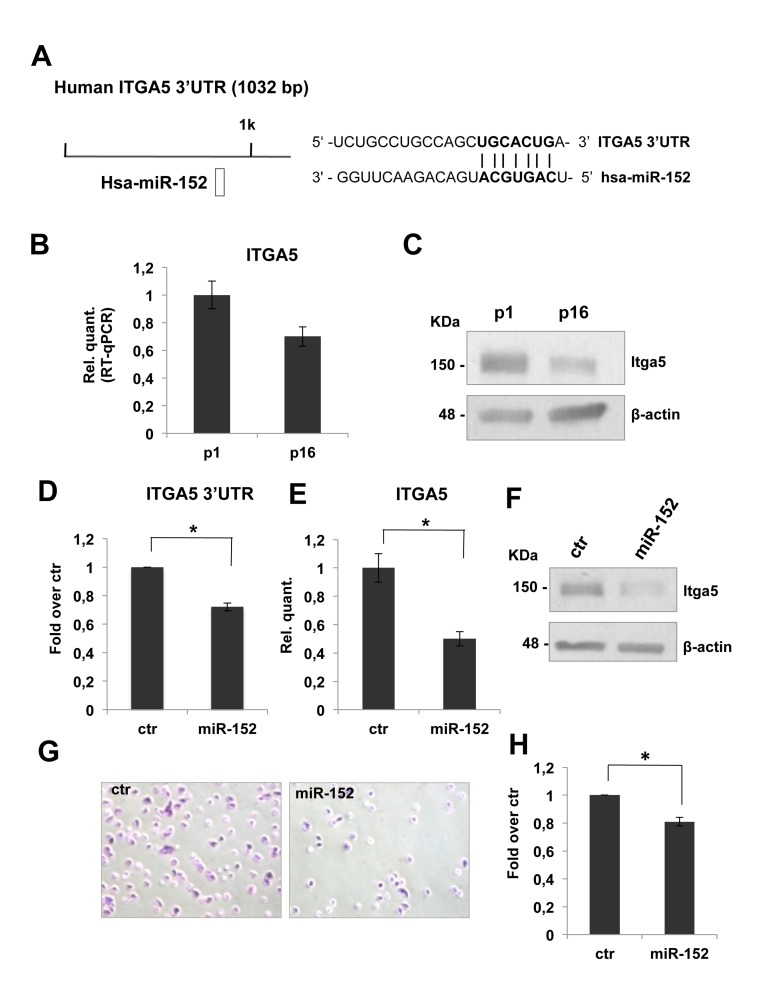
Figure 3. miR-152 represses ITGA5 expression and controls HDFn cell adhesion (A) Predicted miR-152 target sites on human ITGA5 3'UTR were identified by TargetScan 6.2 software. (B) Real Time RT-qPCR was employed to analyze the expression level of ITGA5 in HDFn cells at a growing number of passages in culture (p1-p16). Values reported are the average ± SD of three independent experiment. (C) Western blot of HDFn cells protein extracts collected at increasing passage number in culture (p1-p16). Itga5 protein level is shown and β-actin was used as loading control. (D) Insertion of ITGA5 3'UTR target sequence in a luciferase reporter vector leads to diminished luciferase activity in presence of miR-152 in HEK293 cells 24h after co-transfection. Histograms show the values resulting as the average ± SD from three independent co-transfections.(E) Real Time RT-qPCR was employed to analyze the expression level of ITGA5 in proliferating HDFn cells transfected with miR-152 versus a scrambled control sequence (Ctr). Values reported are the average ± SD of three independent experiment. (F) Western blot analysis of protein extracts of HDFn transfected with miR-152 versus a scrambled control sequence (Ctr). miR-152 overexpression decreases ITGA5 protein levels; β-actin was used as a loading control. (G) Adhesion assay performed on proliferating HDFn cells transfetcted with miR-152 versus scrambled control sequence. (H) Histogram shows adhesion ability of proliferating HDFn 96h after transfection with control or miR-152. Values reported are the average ± SD of three independent experiment. *p-Value <0.01 by Student's t test.
To confirm that ITGA5 mRNA is direct miR-152 target, we cloned ITGA53'UTR sequence, containing the miR-152 conserved binding site, downstream of a luciferase reporter gene. Relative luciferase activity, quantified 24 h after the transfection of reporter construct in presence of pre-miR-152, demonstrated that miR-152 repressed luciferase activity controlled by ITGA5 3-UTR (Figure 3D). Moreover miR-152 transfection in proliferating HDFn is followed by a significant decrease (about 50%, Figure 3E) of ITGA5 mRNA level and led to a strong down-regulation of itga5 protein level (Figure 3F) demonstrating that miR-152 was able to repress HDFn itga5 endogenous expression.
To define the role of miR-152 and its molecular target ITGA5 in HDFn cell adhesion, we tested cell adhesion of proliferating HDFn over-expressing miR-152 compared to scrambled control. We found a decrease of about 20% in cells adhesion in miR-152 transfected cells as compared to control as shown in Figure 3G-H. These results suggested that miR-152 contribute to fibroblast adhesion in part by down regulation of ITGA5.
COL16A1 expression is downregulated in senescent HDFn cells and its 3'-UTR is a direct miR-181a target
To identify potential miR-181a putative targets, we used the TargetScan 6.2 database. COL16A1 was the best candidate target to investigate the possible role of miR-181a on ECM remodeling. Collagen XVI is a member of the fibril associated collagens with interrupted triple helices (FACIT) and constitutes a minor component of the skin ECM. The presence of collagen XVI in the DEJ zone of the papillary dermis indicates an active role in anchoring microfibrils to basement membranes. By interconnecting ECM proteins to cells, collagen XVI is likely to be able to move these proteins and hence affects ECM networks, ensuring mechanical anchorage of the cell and outside-inside signal transduction [85-86].
COL16A1-3'UTR harbours a putative target sequence for miR-181a that is highly conserved among vertebrates (Figure 4A). As shown in figures 4B and 4C, real time-qPCR showed a decrease of COL16A1 mRNA and protein level in dermal fibroblasts from p1 to p16. This inverse correlation with miR-181a expression is consistent with the hypothesis that they might regulate COL16A1. To confirm these, we cloned COL16A1-3'UTR sequence downstream of luciferase cDNA, to use them in luc-reporter assays. Transfecting COL16A1-3'UTR reporter construct in presence of the pre-miR-181a or scrambled sequence, we obtained a significant downregulation, about 30%, in relative luciferase activity (Figure 4D). Overexpression of miR-181a by transfection in proliferating HDFn led to a significant decrease in a COL16A1 mRNA (about 30%, Figure 4E) but not in protein level as shown by western blot analysis in Figure 4F. These results suggested that COL16A1 mRNA is a direct target of miR-181a, we think that we could not detect the decrease at protein level, upon 96h transient transfection, because col16a1 is a very stable protein.
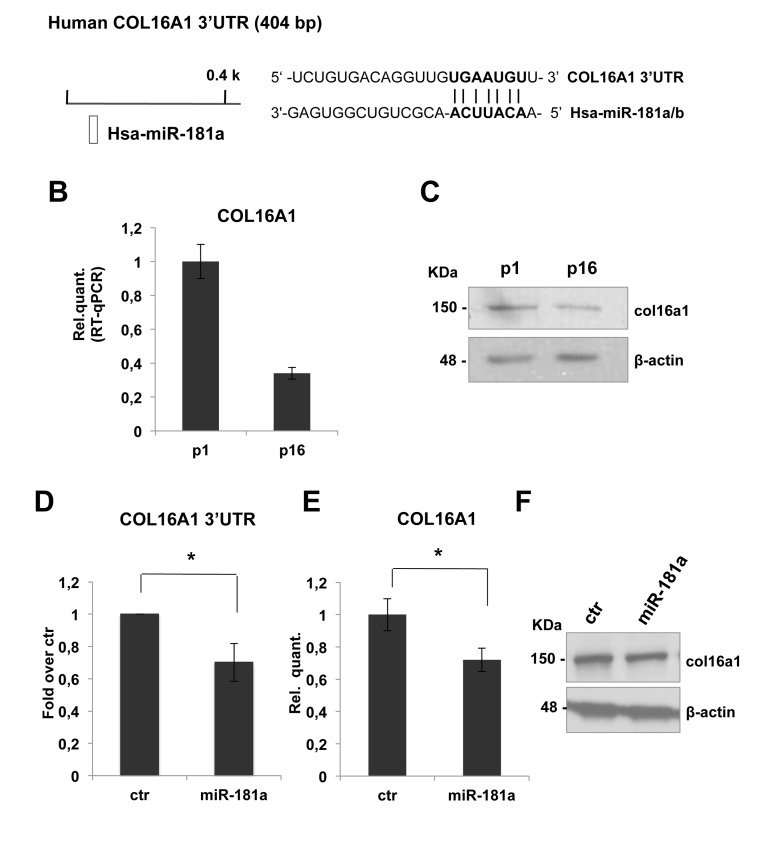
Figure 4. COL16A1 expression is downregulated in senescent HDFn cells and its 3'-UTR is direct miR-181a target (A) Predicted miR-181a target site on human COL16A1 3'UTR was identified by TargetScan 6.2 software. (B) Real Time RT-qPCR was employed to analyze the expression level of COL16A1 in HDFn cells at a growing number of passages in culture (p1-p16). Values reported are the average ± SD of three independent experiment. (C) Western blot of HDFn cells protein extracts collected at increasing passage number in culture (p1-p16). Col16a1 protein level is shown and β-actin was used as loading control. (D) Insertion of COL16A1 3'UTR target sequence in a luciferase reporter vector leads to diminished luciferase activity in presence of miR-181a in HEK293 cells 24h after co-transfection. Histograms show the values resulting as the average ± SD from three independent co-transfections. (E) Real Time RT-qPCR was employed to analyze the expression level of COL16A1 in proliferating HDFn cells transfected with miR-181a versus a scrambled control sequence (ctr). Values reported are the average ± SD of three independent experiment. (F) Western blot analysis of protein extracts of HDFn transfected with miR-181a versus a scrambled control sequence (ctr). Itga5 protein levels is shown and β-actin was used as a loading control.
Discussion
Here, we have identified two miRNAs, miR-152 and miR-181a, upregulated in senescent human diploid fibroblasts. We have also shown that 3-5 fold expression of the two miRNAs is sufficient per se to induce senescence markers (SA-β-gal staining, reduced cell proliferation and expression of p16INK4a) in proliferating, young fibroblasts. We have identified ITGA5 as direct targets of miR-152. Integrins, beside transducing signals from the matrix to cells, bind to ECM to allow the fibroblasts to organize and remodel the matrix [81], this features is very limited in aged tissues and in particular in aged skin and this could be due in part to ITGA5 downregulation mediated by miR-152. In addition, we have shown that overexpression of miR-152 in fibroblasts significantly reduced cell adhesion, this finding indicate that ITGA5 might have a role in aged skin tissue. On the other hand, miR-181a target is COL16A1 mRNA, collagen XVI is a minor component of the skin ECM, nevertheless it is expressed in the DEJ zone of the papillary dermis and it connects ECM proteins to cells, ensuring mechanical anchorage of the cell and outside-inside signal transduction [85, 86]. Overall, these findings suggest a model whereby during replicative senescence, a set of miRNAs, among them miR-152 and miR-181a, are upregulated, this sustain the senescent phenotype. The mechanisms through which the miRNAs are upregulated will deserve further investigation. Although we do not exclude the possibility that other important senescence-associated miRNA-152 and miR-181a targets play a role in the senescent phenotype observed, we believe that the reduction in expression of ITGA5 and COL16A1 by miR-152 and miR-181a, strongly suggests that miRNAs have a complex role also in ECM remodeling typical of aged skin that deserve to be further investigated.
Material and methods
Cell culture and Transfection
Human Dermal Fibroblasts neonatal (HDFn, Cascade, Invitrogen, Carlsbad, California, USA) were cultured in 106 medium added with LSGS growth supplements (Cascade). Cells were passaged usually once a week, at each passage the harvested cells number and seeded cell number were recorded in order to calculate the population doublings occurring between passages and the population doubling time. At each passage different aliquots of the cells were harvested to extract in triplicate RNA and proteins and an aliquot was submitted to senescence activated β-galactosidase staining in order to assay the senescent or non-senescent state of the cells. Human primary fibroblasts were transfected with human pre-miR 181a, pre-miR-152, and scramble sequence as negative control (Ambion, Texas, USA) using the Lipofecatmine RNAimax transfection reagent (Invitrogen) according to manufacturer protocols. 24hrs after transfection, the medium was removed and replaced with fresh medium.
RNA Extraction and Real Time PCR analysis
Total RNA from cells was isolated using mirVana mirRNA Isolation Kit (Ambion) following the manufacturer's protocol. Total RNA was quantified using a NanoDrop Spectophotometer (Thermo Scientific, Delaware, USA) and RNA quality was controlled on an 1% agarose gel. For microRNA detection, RNA was reverse transcribed using TaqMan MicroRNA Reverse Transcription kit and qRT-PCR was performed with TaqMan universal master mix (Applied Biosystem) and specific primers for miR-181a and miR-152. U18 was used as housekeeping gene for normalization (Applied Biosystem).
For determination of mRNA expression level, total RNA was reverse transcribed with GoScriptTM Reverse Transcription System (Promega, Madison, WI, USA) according to manufacturer's protocols. Real Time PCR was than performed with COL16A1 or ITGA5 specific primers by using the Platinum SYBR Green qPCR SuperMix UDG (Invitrogen). The sequences of the primers used in this study were as follow: hCOL16A1F 5' –CTTCAACCTCATCCACCGACTCAG- 3', hCOL 16A1R 5' –TAGTGTCAGCACCAGGGCGGCAAAC TC- 3', hITGA5F 5'-TTGTTGCTGGTGTGCCCAAA G-3', hITGA5R 5'-GCCATCTGTTCCCCTGAGAAG-3'. β-Actin was used as a housekeeping gene for normalization. The expression of each gene and miR was defined from the threshold cycle (Ct), and relative expression levels were calculated by using the 2−ΔΔCt method after normalization with reference to expression of housekeeping genes.
Senescence-Associated β-Galactosidase Staining
Cells were grown in 6-well culture plates, washed with PBS, and fixed with 2% formaldehyde/0.2% glutaral-dehyde/2mM MgCl2 in PBS for 5 minutes. After another washing step with PBS, cells were incubated with β-galactosidase staining solution (2 mM MgCl2, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 1 mg/mL 5-bromo-4-chloro-3-indolyl- β-Dgalactoside [X-gal], pH 6.0) for 24 hours at 37°C. The reaction was stopped by replacing the staining solution with 70% glycerol.
Cell proliferation and cell cycle analysis
Incorporation of bromodeoxyuridine (BrdU) during DNA synthesis was evaluated with the Click-iT™ EdU flow cytometry assay kit, following the manufacturer's protocol (Molecular Probes, Eugene, OR, USA). Cell cycle was analysed using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). Fifteen thousand events were evaluated using the Cell Quest (BD) software.
Luciferase Assay and constructs
The 3'-UTRs of miR-181a, and miR-152 target mRNAs were amplified by PCR from human genomic DNA using the following primer pairs: hCOL16A1-3'UTR-F 5'-GGCCTCTAGA CCCCACCTGCCTTTGGATG -3'; hCOL16A1-3'UTR-R 5'-GGCCTCTAGAGACTGAGTCTCATTA GTTGC -3'; hITGA5-3'UTR-F 5'-GGCCTCTAGAGT CCTCCCAATTTCAGACTC -3'; hITGA5-3'UTR-R 5'-GGCCTCTAGACTAGTTCTGGTCAGTGGGGG -3'. PCR fragments were restricted and ligated to a compatible XbaI-linearized pGL3Control vector (Promega). Cells were transfected with 100ng of pGL3 vectors, 12pmol of pre-miR or a scrambled sequence (Ambion), and10ng of Renilla luciferase pRL-CMVvector (Promega). Luciferase assays were then performed as described before [87].
Adhesion Assay
96-well microplates were coated for 1h at 37°C with 100 μg/ml rat tail collagen I (BD, Franklin Lakes, NJ USA) and blocked for 1h at 37°C with PBS, 1% (W/V) BSA.96h after transfection 1.2 x104 cells were detached, seeded into 96-well culture plate and incubated for 15 minutes at 37°C/5% CO2. The adherent cells were washed twice with PBS, fixed in 4% paraformaldehyde for 15 minutes and stained with 0.1% crystal violet in 2% ethanol for 10 minutes. Excess dye was removed by washing with H2O, the violet stain was solubilized with 2% SDS and absorbance was measured at 540 nm with ELISA reader.
Western Blotting
Total cell extracts were resolved on a SDS polyacrylamide gel, blotted on a Hybond P PVDF membrane (G&E Healthcare, UK). Membranes were blocked with PBST 5% non fat dry milk, incubated with primary antibodies for 2 h at room temperature, washed and hybridized for 1h at room temperature using the appropriate horseradish peroxidase-conjugated secondary antibody (rabbit and mouse, BioRad, Hercules, California, USA). Detection was performed with the ECL chemiluminescence kit (Perkin Elmer, Waltham, Massachusetts, USA). The following antibodies were used: anti-p16 (Santa Cruz Biotechnology, California, USA; dilution 1:1000), anti-col16a1 (Proteintech, Chicago, USA; dilution 1:400), anti-itga5 (Santa Cruz Biotechnology, California, USA; dilution 1:500), anti-p53 (Santa Cruz Biotechnology, California, USA; dilution 1:500), anti-β actin (Sigma, St Louis, Minnesota, USA; dilution 1:5000).
Bioinformatics
Analysis of miR-181A, and miR-152 target sites on COL16A1 and ITGA5 3'UTR were performed using the TargetScan 5.1 software available at http://www.targetscan.org/.
Abbreviations
microRNA: miRNA or miR-; ITGA5: integrin α5; COL16A1: collagen XVI; ECM: extracellular matrix; HDFn: normal human dermal fibroblasts; SA: senescence associated; DEJ: derma-epidermal junctions; BrdU: 5-bromo-2'-deoxyuridine.
Acknowledgments
We thank Dr Anna Maria Lena for scientific discussion. The work reported in this manuscript has been supported by grants from Medical Research Council (UK), MIUR, MinSan, RF73, RF57, ACC12, to GM and by RF73, RF57 and ACC12 to EC.
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88: 593 -602. [PubMed] .
- 2. Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001; 11: S27 -31. [PubMed] .
- 3. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002; 109: 335 -346. [PubMed] .
- 4. Sharpless NE and DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest. 2004; 113: 160 -168. [PubMed] .
- 5. Sommer M, Poliak N, Upadhyay S, Ratovitski E, Nelkin BD, Donehower LA, Sidransky D. DeltaNp63alpha overexpression induces downregulation of Sirt1 and an accelerated aging phenotype in the mouse. Cell Cycle. 2006; 5: 2005 -2011. [PubMed] .
- 6. Campisi J and d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007; 8: 729 -740. [PubMed] .
- 7. Fulco M and Sartorelli V. Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle. 2008; 7: 3669 -79. [PubMed] .
- 8. Tavana O, Benjamin CL, Puebla-Osorio N, Sang M, Ullrich SE, Ananthaswamy HN, Zhu C. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle. 2010; 9: 3328 -3336. [PubMed] .
- 9. Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003; 113: 703 -716. [PubMed] .
- 10. Sharpless NE. Ink4a/Arf links senescence and aging. Exp Gerontol. 2004; 39: 1751 -1759. [PubMed] .
- 11. Ryu SJ, Oh YS, Park SC. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 2007; 14: 1020 -1028. [PubMed] .
- 12. Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, Pilpel Y, Nielsen FC, Oren M, Lund AH. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010; 17: 236 -245. [PubMed] .
- 13. Castro ME, del Valle Guijarro M, Moneo V, Carnero A. Cellular senescence induced by p53-ras cooperation is independent of p21waf1 in murine embryo fibroblasts. J Cell Biochem. 2004; 92: 514 -524. [PubMed] .
- 14. Müller KC, Welker L, Paasch K, Feindt B, Erpenbeck VJ, Hohlfeld JM, Krug N, Nakashima M, Branscheid D, Magnussen H, Jörres RA, Holz O. Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir Res. 2006; 21: 7 32 .
- 15. Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007; 10: 61 -74. [PubMed] .
- 16. Marcel V, Dichtel-Danjoy ML, Sagne C, Hafsi H, Ma D, Ortiz-Cuaran S, Olivier M, Hall J, Mollereau B, Hainaut P, Bourdon JC. Biological functions of p53 isoforms through evolution: lessons from animal and cellular models. Cell Death Differ. 2011; 18: 1815 -1824. [PubMed] .
- 17. Nakamura T and Lipton SA. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011; 18: 1478 -1486. [PubMed] .
- 18. Hendel A, Hiebert PR, Boivin WA, Williams SJ, Granville DJ. Granzymes in age-related cardiovascular and pulmonary diseases. Cell Death Differ. 2010; 17: 596 -606. [PubMed] .
- 19. Darrah E and Rosen A. Granzyme B cleavage of autoantigens in autoimmunity. Cell Death Differ. 2010; 17: 624 -632. [PubMed] .
- 20. Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. 2010; 17: 616 -623. [PubMed] .
- 21. Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011; 18: 9 1487 -1499. [PubMed] .
- 22. Pinton P, Giorgi C, Pandolfi PP. The role of PML in the control of apoptotic cell fate: a new key player at ER-mitochondria sites. Cell Death Differ. 2011; 18: 1450 -6. [PubMed] .
- 23. Hofius D, Munch D, Bressendorff S, Mundy J, Petersen M. Role of autophagy in disease resistance and hypersensitive response-associated cell death. Cell Death Differ. 2011; 18: 1257 -1262. [PubMed] .
- 24. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011; 18: 571 -580. [PubMed] .
- 25. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011; 18: 581 -588. [PubMed] .
- 26. Liang C. Negative regulation of autophagy. Cell Death Differ. 2010; 17: 1807 -1815. [PubMed] .
- 27. Geering B and Simon HU. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. 2011; 18: 1457 -1469. [PubMed] .
- 28. Kelly PN and Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011; 18: 1414 -1424. [PubMed] .
- 29. Füllgrabe J, Hajji N, Joseph B. Cracking the death code: apoptosis-related histone modifications. Cell Death Differ. 2010; 17: 1238 -1243. [PubMed] .
- 30. Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005; 6: 328 -340. [PubMed] .
- 31. Candi E, Rufini A, Terrinoni A, Dinsdale D, Ranalli M, Paradisi A, De Laurenzi V, Spagnoli LG, Catani MV, Ramadan S, Knight RA, Melino G. Differential roles of p63 isoforms in epidermal development: selective genetic complementation in p63 null mice. Cell Death Differ. 2006; 13: 1037 -1047. [PubMed] .
- 32. Núñez R, Sancho-Martínez SM, Novoa JM, López-Hernández FJ. Apoptotic volume decrease as a geometric determinant for cell dismantling into apoptotic bodies. Cell Death Differ. 2010; 17: 1665 -1671. [PubMed] .
- 33. Royer C and Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 2011; 18: 1470 -1407. [PubMed] .
- 34. Smertenko A and Franklin-Tong VE. Organisation and regulation of the cytoskeleton in plant programmed cell death. Cell Death Differ. 2011; 18: 1263 -1270. [PubMed] .
- 35. Leadsham JE, Kotiadis VN, Tarrant DJ, Gourlay CW. Apoptosis and the yeast actin cytoskeleton. Cell Death Differ. 2010; 17: 754 -762. [PubMed] .
- 36. Carmona-Gutierrez D, Eisenberg T, Büttner S, Meisinger C, Kroemer G, Madeo F. Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ. 2010; 17: 763 -773. [PubMed] .
- 37. El-Domyati M, Attia S, Saleh F, Brown D, Birk DE, Gasparro F, Ahmad H, Uitto J. Intrinsic aging vs. photoaging: a comparative histopathological, immunohistochemical, and ultrastructural study of skin. Exp Dermatol. 2002; 11: 398 -405. [PubMed] .
- 38. Lavker RM, Zheng PS, Dong G. Aged skin: a study by light, transmission electron, and scanning electron microscopy. J Invest Dermatol. 1987; 88: 44s -51s. [PubMed] .
- 39. Carrino DA, Onnerfjord P, Sandy JD, Cs-Szabo G, Scott PG, Sorrell JM, Heinegård D, Caplan AI. Age-related changes in the proteoglycans of human skin. Specific cleavage of decorin to yield a major catabolic fragment in adult skin. J Biol Chem. 2003; 278: 17566 -17572. [PubMed] .
- 40. Varani J. Epithelial cell invasion of the stroma in human skin organ culture. Front Biosci. 2004; 9: 2989 -2995. [PubMed] .
- 41. Varani J, Dame MK, Rittie L, Fligiel SE, Kang S, Fisher GJ, Voorhees JJ. Decreased collagen production in chronologically aged skin: roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006; 168: 1861 -1868. [PubMed] .
- 42. Fisher GJ, Quan T, Purohit T, Shao Y, Cho MK, He T, Varani J, Kang S, Voorhees JJ. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am J Pathol. 2009; 174: 101 -114. [PubMed] .
- 43. Goldstein S. Replicative senescence: the human fibroblast comes of age. Science. 1990; 249: 1129 -1133. [PubMed] .
- 44. Cristofalo VJ and Pignolo RJ. Replicative senescence of human fibroblast-like cells in culture. Physiol Rev. 1993; 73: 617 -638. [PubMed] .
- 45. Manning JA and Kumar S. A potential role for NEDD1 and the centrosome in senescence of mouse embryonic fibroblasts. Cell Death Dis. 2010; 1: e35 [PubMed] .
- 46. Gonzalez-Cano L, Herreros-Villanueva M, Fernandez-Alonso R, Ayuso-Sacido A, Meyer G, Garcia-Verdugo JM, Silva A, Marques MM, Marin MC. p73 deficiency results in impaired self renewal and premature neuronal differentiation of mouse neural progenitors independently of p53. Cell Death Dis. 2010; 1: e109 [PubMed] .
- 47. Giampietri C, Petrungaro S, Coluccia P, Antonangeli F, Giannakakis K, Faraggiana T, Filippini A, Cossu G, Ziparo E. c-Flip overexpression affects satellite cell proliferation and promotes skeletal muscle aging. Cell Death Dis. 2010; 1: e38 [PubMed] .
- 48. Drullion C, Trégoat C, Lagarde V, Tan S, Gioia R, Priault M, Djavaheri-Mergny M, Brisson A, Auberger P, Mahon FX, Pasquet JM. Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell Death Dis. 2012; 3: e373 [PubMed] .
- 49. Schneider EL. Aging and cultured human skin fibroblasts. J Invest Dermatol. 1979; 73: 15 -18. [PubMed] .
- 50. Hayflick L. The cell biology of aging. J Invest Dermatol. 1979; 73: 8 -14. [PubMed] .
- 51. West MD, Pereira-Smith OM, Smith JR. Replicative senescence of human skin fibroblasts correlates with a loss of regulation and overexpression of collagenase activity. Exp Cell Res. 1989; 184: 138 -147. [PubMed] .
- 52. Vilborg A, Bersani C, Wilhelm MT, Wiman KG. The p53 target Wig-1: a regulator of mRNA stability and stem cell fate? Cell Death Differ. 2011; 18: 9 1434 -40. [PubMed] .
- 53. Garofalo M, Condorelli GL, Croce CM, Condorelli G. MicroRNAs as regulators of death receptors signaling. Cell Death Differ. 2010; 17: 2 200 -208. [PubMed] .
- 54. Yi R and Fuchs E. MicroRNA-mediated control in the skin. Cell Death Differ. 2010; 17: 229 -35. [PubMed] .
- 55. Godlewski J, Newton HB, Chiocca EA, Lawler SE. MicroRNAs and glioblastoma; the stem cell connection. Cell Death Differ. 2010; 17: 221 -228. [PubMed] .
- 56. Aqeilan RI, Calin GA, Croce CM. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 2010; 17: 215 -220. [PubMed] .
- 57. Lund AH. miR-10 in development and cancer. Cell Death Differ. 2010; 17: 209 -214. [PubMed] .
- 58. Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010; 17: 193 -199. [PubMed] .
- 59. Grillari J and Grillari-Voglauer R. Novel modulators of senescence, aging, and longevity: Small non-coding RNAs enter the stage. Exp Gerontol. 2010; 45: 302 -311. [PubMed] .
- 60. Hackl M, Brunner S, Fortschegger K, et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell. 2010; 9: 291 -296. [PubMed] .
- 61. Faraonio R, Pane F, Intrieri M, Russo T, Cimino F. In vitro acquired cellular senescence and aging-specific phenotype can be distinguished on the basis of specific mRNA expression. Cell Death Differ. 2002: 9: 862 -86. [PubMed] .
- 62. Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Quirantes R, Segura-Carretero A, Micol V, Joven J, Bosch-Barrera J, Del Barco S, Martin-Castillo B, Vellon L, Menendez JA. Metformin lowers threshold for stress-induced senescence: a role for microRNA-200 family and niR-205. Cell Cycle. 2012; 1: 1235 -1246. .
- 63. Telgenhoff D and Shroot B. Cellular senescence mechanisms in chronic wound healing. Cell Death Differ. 2005; 12: 695 -698. [PubMed] .
- 64. Marasa BS, Srikantan S, Martindale JL, Kim MM, Lee EK, Gorospe M, Abdelmohsen K. MicroRNA profiling in human diploid fibroblasts uncovers miR-519 role in replicative senescence. Aging (Albany NY). 2010; 2: 333 -343. [PubMed] .
- 65. He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network—another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007; 7: 819 -822. [PubMed] .
- 66. Lafferty-Whyte K, Cairney CJ, Jamieson NB, Oien KA, Keith WN. Pathway analysis of senescence-associated miRNA targets reveals common processes to different senescence induction mechanisms. Biochim Biophys Acta. 2009; 1792: 341 -352. [PubMed] .
- 67. Maes OC, Sarojini H, Wang E. Stepwise up-regulation of microRNA expression levels from replicating to reversible and irreversible growth arrest states in WI-38 human fibroblasts. J Cell Physiol. 2009; 221: 109 -119. [PubMed] .
- 68. Saunders LR, Sharma AD, Tawney J, Nakagawa M, Okita K, Yamanaka S, Willenbring H, Verdin E. miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging (Albany NY). 2010; 2: 415 -431. [PubMed] .
- 69. Jun JI and Lau LF. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY). 2010; 2: 627 -631. [PubMed] .
- 70. Gorospe M and Abdelmohsen K. MicroRegulators come of age in senescence. Trends Genet. 2011; 27: 233 -241. [PubMed] .
- 71. Smith SM and Murray DW. An overview of microRNA methods: expression profiling and target identification. Methods Mol Biol. 2012; 823: 119 -138. [PubMed] .
- 72. Fink LS, Roell M, Caiazza E, Lerner C, Stamato T, Hrelia S, Lorenzini A, Sell C. 53BP1 contributes to a robust genomic stability in human fibroblasts. Aging (Albany NY). 2011; 3: 836 -845. [PubMed] .
- 73. Mao Z, Ke Z, Gorbunova V, Seluanov A. Replicatively senescent cells are arrested in G1 and G2 phases. Aging (Albany NY). 2012; 4: 431 -435. [PubMed] .
- 74. Lena AM, Mancini M, Rivetti di Val Cervo P, Saintigny G, Mahé C, Melino G, Candi E. MicroRNA-191 triggers keratinocytes senescence by SATB1 and CDK6 downregulation. Biochem Biophys Res Commun. 2012; 423: 509 -514. [PubMed] .
- 75. Rivetti di Val Cervo P, Lena AM, Nicoloso M, Rossi S, Mancini M, Zhou H, Saintigny G, Dellambra E, Odorisio T, Mahé C, Calin GA, Candi E, Melino G. p63-microRNA feedback in keratinocyte senescence. Proc Natl Acad Sci USA. 2012; 109: 1133 -118. [PubMed] .
- 76. Wang Y, Scheiber MN, Neumann C, Calin GA, Zhou D. MicroRNA regulation of ionizing radiation-induced premature senescence. Int J Radiat Oncol Biol Phys. 2011; 81: 839 -848. [PubMed] .
- 77. Dhahbi JM, Atamna H, Boffelli D, Magis W, Spindler SR, Martin DI. Deep sequencing reveals novel microRNAs and regulation of microRNA expression during cell senescence. PLoS One. 2011; 6: e20509 [PubMed] .
- 78. Faraonio R, Salerno P, Passaro F, Sedia C, Iaccio A, Bellelli R, Nappi TC, Comegna M, Romano S, Salvatore G, Santoro M, Cimino F. A set of miRNAs participates in the cellular senescence program in human diploid fibroblasts. Cell Death Differ. 2012; 19: 713 -721. [PubMed] .
- 79. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002; 110: 673 -687. [PubMed] .
- 80. Luo BH and Springer TA. Integrin structures and conformational signaling. Curr Opin Cell Biol. 2006; 18: 579 -586. [PubMed] .
- 81. Leiss M, Beckmann K, Girós A, Costell M, Fässler R. The role of integrin binding sites in fibronectin matrix assembly in vivo. Curr Opin Cell Biol. 2008; 20: 502 -507. [PubMed] .
- 82. Desgrosellier JS and Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010; 10: 9 -22. [PubMed] .
- 83. Watt FM and Hodivala KJ. Cell adhesion. Fibronectin and integrin knockouts come unstuck. Curr Biol. 1994; 4: 270 -272. [PubMed] .
- 84. Wang QY, Zhang Y, Shen ZH, Chen HL. alpha1,3 fucosyltransferase-VII up-regulates the mRNA of alpha5 integrin and its biological function. J Cell Biochem. 2008; 104: 2078 -2090. [PubMed] .
- 85. Grässel S, Unsöld C, Schäcke H, Bruckner-Tuderman L, Bruckner P. Collagen XVI is expressed by human dermal fibroblasts and keratinocytes and is associated with the microfibrillar apparatus in the upper papillary dermis. Matrix Biol. 1999; 18: 309 -317. [PubMed] .
- 86. Kassner A, Hansen U, Miosge N, Reinhardt DP, Aigner T, Bruckner-Tuderman L, Bruckner P, Grässel S. Discrete integration of collagen XVI into tissue-specific collagen fibrils or beaded microfibrils. Matrix Biol. 2003; 22: 131 -143. [PubMed] .
- 87. Lena AM, Shalom-Feuerstein R, Rivetti di Val Cervo P, Aberdam D, Knight RA, Melino G, Candi E. miR-203 represses ‘stemness’ by repressing DeltaNp63. Cell Death Differ. 2008; 15: 1187 -1195. [PubMed] .