



Introduction
Increased levels of reactive oxygen species (ROS) are often associated with microvascular pathology in age-related cardiovascular diseases (CVD) including coronary artery disease (CAD) and ischemic heart disease (IHD) - the major causes of death and morbidity in the USA[1-3]. These observations have led to the current paradigms in the field of cardiovascular and stroke research that reduction in ROS levels in the vessel walls should improve vascular functions [4]. However, interventional clinical trials using global antioxidants, e.g. HOPE, ATBC [5-8], have largely produced negative results in reducing primary endpoints of cardiovascular death and morbidity (with some suggestion of potential harm). Recent studies by us and others using genetically manipulated animal models and/or antioxidants in cultured endothelial cells (ECs) demonstrated that reduction in ROS did not improve vascular functions[9-12]. Instead, reduced ROS levels resulted in the disruption of the signal transduction events that are essential for nitric oxide (NO) generation in vascular endothelium[10, 13-15], which in turn impaired vasodilatation[10, 14, 16]. Together, these findings suggest that increased ROS levels often present in elderly patients with CVD may play critical roles in endothelial function.
Precise mechanisms by which ROS modulate signal transduction in ECs are not fully understood. Several studies have shown that ROS play critical roles in ECs by activating signaling intermediates including PI3K-Akt-eNOS, PLCγ1, PKC and ERK1/2 [9, 17, 18]. Recent reports suggest that the pro-survival kinase AMPK, which becomes activated during cellular stress including caloric restriction and reduction in AMP/ATP ratio, can also be regulated by oxidants generated during hypoxic or fluid shear stress in ECs [19-21]. AMPK, in turn, is known to regulate a number of signaling intermediates and transcription factors including HIF-1α, FOXO1 and PGC-1α [22, 23], resulting in increased EC survival and proliferation. One of the major protective roles of AMPK in cell survival is carried out through its regulatory role in autophagy [21, 24], a cellular process for the recycling and re-utilization of the damaged macromolecules and organelles using intracellular lysosomal degradative pathway [24, 25]. However, it is not known whether ROS regulate activity of the survival kinase AMPK and autophagy in ECs.
There are several sources for intracellular ROS in EC including NADPH oxidases, mitochondria, cytochrome P450 and xanthine oxidase. Rac1-dependent NADPH oxidase is a major source of endothelial ROS, which is a multi-subunit enzyme complex containing membrane-bound gp91phox (Nox2) and p22phox subunits, and cytosolic p47phox, p67phox and Rac1[12, 26, 27]. Several other NADPH oxidases are present in ECs, e.g. nox4, nox1, nox5 [11, 28-31]. Nox2 containing NADPH oxidase is present in different subcellular compartments in ECs including cell and perinuclear membrane, and endoplasmic reticulum (ER)[27, 32]. Recent work from others and our labs has shown a previously unappreciated role for NADPH oxidase-derived ROS in the activation of downstream eNOS to synthesize NO[9-11, 14, 15, 33, 34]. These studies suggest that NADPH oxidase-derived ROS play a critical positive role in vascular endothelial survival, health and growth. However, several of these studies were performed using cultured ECs in vitro, global knockdown animal models of NADPH oxidase (p47phox−/− or gp91phox−/−), or constitutive over-expression of NADPH oxidases (e.g. Nox4) in ECs[9, 10, 12, 14, 16, 35, 36]. These approaches, although yielding important information, may have precluded precise determination of endothelial contribution for redox-sensitive regulation of vascular functions.
To examine endothelial responses to conditional in vivo increase in EC-specific endogenous ROS in adult animals, we have generated a novel binary (Tet-ON/OFF) transgenic mouse that induces EC-specific 1.8±0.42-fold increase in Nox2/gp91phox (NADPH oxidase 2)-derived ROS for four to 12 weeks. Using isolated mouse heart EC (MHEC) and coronary microvessels, we demonstrate that conditional increase in EC-specific ROS induces AMPK-eNOS-mediated endothelium-dependent coronary vasodilatation, and AMPK-mTOR-mediated protective autophagy in MHEC.
Results
Nox2 overexpression in the vascular endothelium
The double/binary transgenic NVF (TRE-NOX2:VE-Cadherin-tTA) animals were supplied with tetracycline in the drinking water to turn off the transgene expression as described in the Materials and Methods (Fig. 1A). Withdrawal of tetracycline from the drinking water (Tet-OFF) for at least 2 weeks induced NOX2-HA specifically in the endothelium of the double transgenic mice (Fig. 1B-1C, shows Tet-OFF for 4 weeks. Supplemental Fig. 1A shows Tet-OFF for 8 weeks). In order to examine the effects of Nox2 overexpression in ECs, mouse heart endothelial cells (MHEC) were isolated from two independent binary transgenic mouse lines as described [10]. MHECs from Tet-ON animals were grown in medium containing tetracycline (2 μg/ml) and Tet-OFF MHEC were grown in medium without tetracycline. Western blots using protein lysates and RT-PCR using RNA from Tet-OFF MHEC demonstrated significantly increased levels of Nox2 protein and Nox2 mRNA, respectively, compared to Tet-ON MHEC (Fig. 1D-1E). To determine whether EC-specific Nox2 overexpression increased ROS levels, DCF-DA fluorescence assays using FACS were performed. Tet-OFF MHEC showed 1.8±0.42-fold increase in ROS levels compared to Tet-ON MHEC (Fig. 1F), showing that overexpression of Nox2 resulted in increased ROS levels in ECs. However, there were no significant changes in the expression levels of other components of NADPH oxidase complex tested such p47phox, p22phox or nox4 (data not shown). Si-RNA knockdown of Nox2 in Tet-OFF MHEC resulted in significant inhibition of NADPH oxidase activity (Supplemental Fig. 1B) and decrease in ROS levels (Supplemental Fig. 1C), suggesting that increased ROS levels observed in Tet-OFF MHEC were due to increase in Nox2-mediated NADPH oxidase activity. In order to confirm specificity of DCF-fluorescence by ROS, catalase but not L-NAME was shown to inhibit ROS in Tet-OFF MHEC (Supplemental Fig. 1D). Together, these data demonstrate that the binary transgenic mouse (NVF) can generate increased levels of endogenous ROS in ECs upon withdrawal of tetracycline.
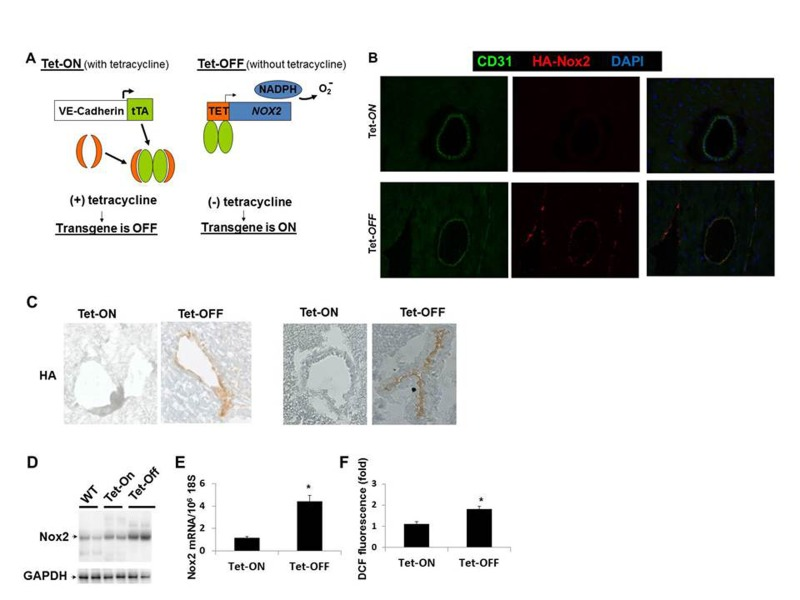
Figure 1. Endothelium-specific overexpression of Nox2 and ROS generation (A) Schematic used to make conditional binary transgenic mice (NVF). Tet-ON, tetracycline in the drinking water to suppress the transgene; Tet-OFF, withdrawal of tetracycline for four weeks to induce the transgene. (B) Frozen heart sections of 8 weeks old Tet-ON and Tet.OFF NVF (for 4 weeks) mice showing EC-specific (CD31, green) HA-tagged Nox2 (red) expression in coronary vessels of Tet-OFF animal. (C) Immunohistochemistry using anti-HA antibody on frozen heart sections. (D) Western blots using transgenic mouse heart EC (MHEC) lysates showing Nox2 overexpression in two independent lines of Tet-OFF animals compared to Tet-On and WT animals. (E) Q-PCR using MHEC RNAs from Tet-ON and Tet-OFF animals (n=6/group). (F) FACS analyses using DCF fluorescence of MHECs (n=6/group). All animals were 8 weeks old, Tet-OFF were without tetracycline for four weeks. Tet-ON MHECs were grown in medium containing tetracycline 2μg/mL. *p<0.05.
Increased coronary vasorelaxation in Tet-OFF mice
The established dogma or classical belief is that ROS reduce bioavailability of NO by converting it to peroxynitrite (ONOO) and thus inhibit endothelium-dependent vasorelaxation. There are, however, conflicting reports about the roles of ROS in vascular biology. Recent findings by others [37] and us [10] demonstrated that NADPH oxidase-derived ROS are essential for endothelium-dependent vasorelaxation. Failure of the clinical trials using antioxidants to improave the health of cardiovascular patients also suggested a critical role for ROS in vascular function [5-8]. To determine whether EC-ROS resulted in the improvement of coronary vasorelaxation or not, we isolated coronary microvessels from Tet-ON and Tet-OFF NVF transgenic mice (8-12 weeks old) and subject endothelium-dependent microvessel reactivity assays using vascular endothelial growth factor (VEGF) and acetylcholine (Ach) as described [10]. Tet-OFF coronary vessels showed significantly increased (by >22%) vasodilation over Tet-ON vessels in response to VEGF and Ach, however, there was no difference in SNP induced relaxation in these vessels (Fig. 2), suggesting that increased ROS levels in EC resulted in endothelium-dependent vasodilatation in coronary vessels. Non-specific effects of tetracycline on Tet-ON animals were excluded by performing microvessel reactivity assays on coronary vessels from WT animals that were treated with or without tetracycline for eight weeks (Supplemental Fig. 2A). Endothelium denudation and eNOS-inhibitor L-NAME (300 μmol/L) inhibited microvessel reactivity in both Tet-ON and Tet-OFF coronary vessels (data not shown), suggesting endothelium-generated NO plays a major role in coronary vasorelaxation. Inhibition of vasodilatation in Tet-OFF animals by another NO-scavenger carboxy-PTIO (200 μmol/L) (Supplemental Fig. 2B), further supported this notion. Supplementation of drinking water with tetracycline in Tet-OFF mice for 4 weeks (Tet-OFF+Tet-ON) and incubation of coronary vessels with ROS-scavenger NAC reduced vasodilation in Tet-OFF animals to the level of that of Tet-ON mice (Supplemental Fig. 2B), suggesting specific role for NADPH oxidase-derived inducible ROS in vasodilatation in these binary transgenic animals. One may wonder whether increased level of ROS and NO resulted in higher ONOO− levels in Tet-OFF vessels, which in turn was responsible for increased vasorelaxation. To address this, microvessel reactivity assays were performed using nitric oxide-cGMP inhibitor, ODQ (10 μmol/L). ODQ inhibited vasodilatation in both Tet-ON and Tet-OFF coronary arterioles (Fig. 2D). Together with c-PTIO data (Supplemental Fig. 2B), these findings suggest that NO, but not ONOO−, plays a major role in EC-ROS-mediated increase in vasorelaxation.
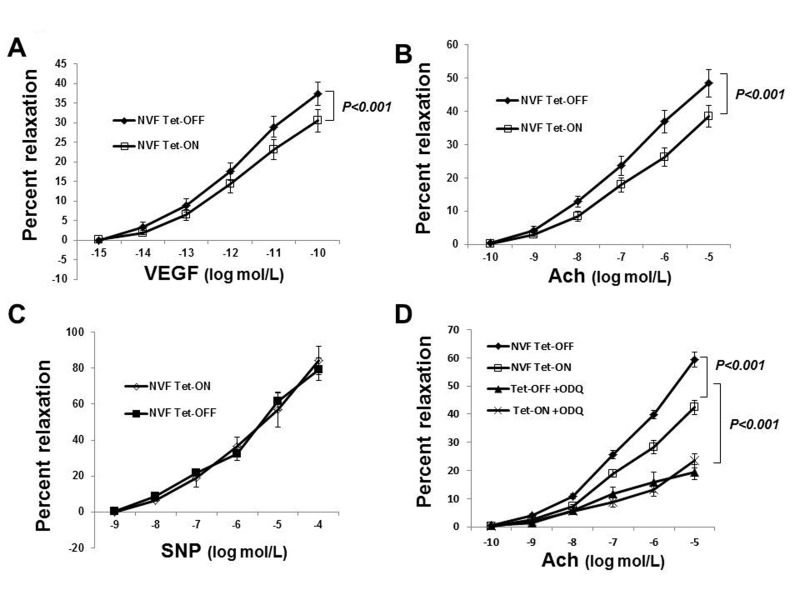
Figure 2. Increased coronary vasodilatation in Tet-OFF mice with higher EC-ROS (A) Endothelium-dependent dilation of coronary arterioles from Tet-ON (n=6) and Tet-OFF (n=6) NVF mice in response to VEGF. 22±3.72% increase in vasodilation in Tet-OFF vs. Tet-ON. (B) Endothelium-dependent dilation of coronary arterioles from Tet-ON (n=6) and Tet-OFF (n=6) NVF mice in response to acetylcholine (Ach). 25±2.43% increased dilation in Tet-OFF vs. Tet-ON. (C) Endothelium-independent dilation of coronary arterioles from Tet-ON (n=6) and Tet-OFF (n=6) mice in response to NO donor, SNP. (D) NO-cGMP signaling inhibitor ODQ (10 μmol/L) inhibited coronary vasorelaxation in both Tet-ON and Tet-OFF coronary vessels. n = 6/group. All coronary vessels were pre-constricted ex-vivo using U46619 prior to the addition of VEGF, Ach or SNP as indicated.
Sustained increase in EC-ROS induces eNOS activation by AMPK
In order to examine the effects of increased ROS on eNOS activation and NO bioavailability, Tet-ON and Tet-OFF MHEC lysates were subject to Western blot analysis. Tet-OFF MHEC demonstrated increased phosphorylation of eNOS (Ser1179) in an AMPK-dependent manner (Fig. 3A-3B). However, there was no difference in total eNOS expression between Tet-ON and Tet-OFF MHEC (Supplemental Fig. 3). AMPK-dependent eNOS activation by EC-ROS was further confirmed by demonstrating increased NO levels in Tet-OFF MHEC and its inhibition by si-AMPK, as determined by citrulline assay (Fig. 3C). Akt phospho-rylation remained unchanged (Fig. 3A). Together, these fidnings suggest increase in EC-ROS results in activation of eNOS in an AMPK-dependent mechanism.
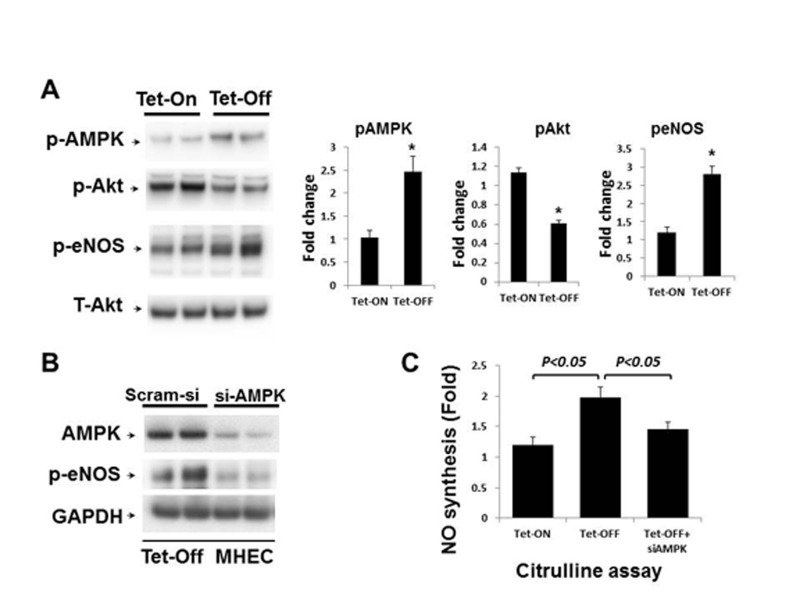
Figure 3. Above-physiological ROS levels induce AMPK-mediated eNOS activation in Tet-OFF MHEC (A) Western blots (WB) analyses of MHEC protein lysates from two independent lines of NVF Tet-ON and Tet-OFF mice as indicated. WB was carried out using anti-phospho-AMPK (p-AMPK), anti-p-Akt (ser473), anti-p-eNOS (ser1179) and anti-T-Akt (total) antibodies. T-Akt was used as loading control. Right panels, bar graphs show quantitative densitometric analysis of three independent experiments using NIH image J (-fold change expressed in mean ± S.E.M.). *p<0.05 was considered statistically significant. (B) Protein extracts from Tet-OFF MHEC transfected with control siRNA (Scram-si) or si-AMPK were subject to Western blots as described in the Methods. Membranes were sequentially blotted, stripped and re-probed with anti-AMPK, anti-p-eNOS and GAPDH antibodies as shown. Representative blots of two independent experiments are shown. (C) NO production, as measured using citruline assay as described in Methods, was 2.1±0.32-fold higher in Tet-OFF MHEC compared to Tet-ON. Si-AMPK significantly inhibited NO production in Tet-OFF MHEC. *p<0.05.
Increased coronary vasorelaxation in Tet-OFF mice is AMPK-dependent
Since eNOS activation in Tet-OFF MHEC is AMPK-dependent, we wanted to examine whether the increase in coronary vasorelaxation in Tet-OFF NVF mice was AMPK-mediated. To that end, isolated coronary vessels from Tet-ON and Tet-OFF transgenic mice were subject to microvessel reactivity assay in the presence or absence of AMPK inhibitor, Compound C (80 μmol/L). Ach- and VEGF-mediated coronary vasodilatation was inhibited by Compound C in Tet-OFF mice, whereas Compound C had no significant effect on the coronary vessels from Tet-ON mice (Fig.4A shows Ach-mediated microvessel reactivity). These results suggest that the observed increase in Tet-OFF coronary vasodilatation was, at least in part, induced by an AMPK-mediated endothelium-dependent signaling pathway.
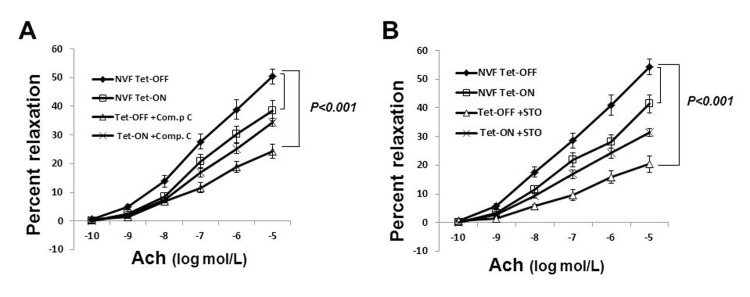
Figure 4. Inhibition of AMPK signaling reduced coronary vasodilatation in Tet-OFF NVF but not in Tet-ON NVF (A) Isolated coronary vessels from Tet-ON and Tet-OFF transgenic mice (n=6/group) were subject to microvessel reactivity assay in the presence or absence of Compound C (80 μmol/L). Ach-mediated vasodilatation was inhibited by Compound C in the coronary vessels from Tet-OFF mice, whereas Compound C had no significant effect on the coronary vessels from Tet-ON mice. (B) Same as in (A), except pre-treatment was carried out using CaMKKβ-inhibitor STO-609 (50 nmol/L).
ROS-induced AMPK-eNOS Activation is CaMKKβ-dependent
Earlier studies demonstrated that CaMKK acts as an upstream kinase for AMPK in ECs. Since ROS-stimulated eNOS activation was AMPK-dependent, we hypothesized that CaMKKβ was likely to be the upstream kinase involved in AMPK activation. To examine selective role of CaMKKβ in ROS-induced AMPK activation, CaMKKβ was knocked down by si-CaMKKβ. Transfection with si-CaMKKβ significantly reduced AMPK Thr-172 phosphorylation in Tet-OFF MHEC (Supplemental Fig. 4), suggesting that AMPK activation by ROS is CaMKKβ-dependent. STO-609 (50 nmol/L) is a potent inhibitor of CaMKK (both CaMKKα and CaMKKβ). This low concentration of STO-609 was shown to be selective for CaMKK, and did not inhibit other kinases including AMPK. STO-609 inhibited endothelium-dependent vasorelaxation in Tet-OFF mouse coronary arterioles (Fig. 4B), suggesting that ROS-induced AMPK-eNOS-mediated vasodilatation is CaMKK-dependent.
Increased EC-ROS inhibit mTOR-p70S6K signaling
AMPK activation is known to have inhibitory effects on mTOR signaling[38, 39]. Since Tet-OFF MHEC with higher levels of ROS demonstrated increased activation of AMPK, we examined whether downstream mTOR/p70S6K/eIF-4E-BP signaling were also affected. Western blots analysis demonstrated marked reduction in mTOR/p70S6K signaling in MHEC with higher ROS (Fig. 5A). Reduction in mTOR signaling was further confirmed by increased levels of 4E-BP, a protein which is degraded by mTOR-mediated phosphorylation, in Tet-OFF MHEC (Fig. 5A). Together, these results suggest that higher levels of NADPH oxidase-derived ROS inhibit mTOR signaling pathway in ECs.
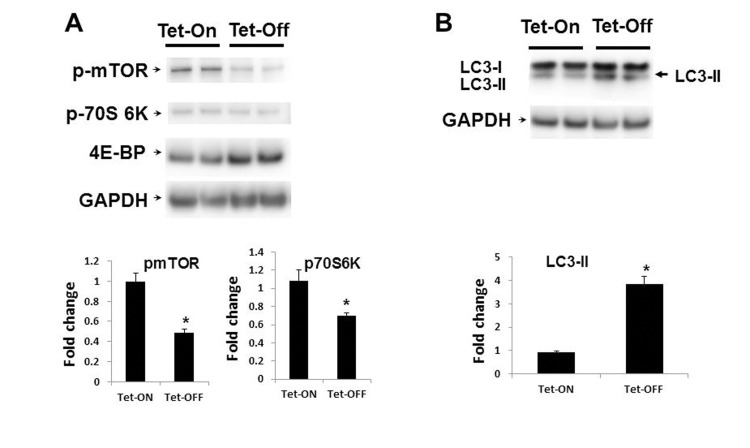
Figure 5. Inhibition of mTOR signaling in Tet-OFF MHEC with high ROS (A) Western blot analyses of MHEC protein lysates from two independent lines of NVF Tet-ON and Tet-OFF mice as indicated. WB was carried out using anti-p-mTOR (p-mTOR), anti-p-70S 6k, and anti-4E-BP antibodies. GAPDH was used for loading control. Lower panels, bar graphs show quantitative densitometric analysis of three independent experiments of the p-mTOR and p-70S 6K bands (-fold change expressed in mean ± S.E.M.). *p<0.05 was considered statistically significant. (B) WB analyses of MHEC from two independent lines of NVF Tet-ON and Tet-OFF as in (A) except anti-LC3A (I and II) and ant-GAPDH antibodies were used. Arrow, induction of the autophagy marker LC3-II in Tet-OFF MHEC is indicated. Lower panel, bar graph showing quantitative analyses of LC3-II as indicated. *p<0.05.
Increased ROS induce autophagy in EC
Since MHEC with higher ROS levels showed activation of AMPK and inhibition of mTOR signaling pathways, we hypothesized that Tet-OFF MHEC would demonstrate increased autophagy. Western blots showed a significant increase in the autophagosome-bound LC3A-II in Tet-OFF MHEC compared to Tet-ON MHEC (Fig. 5B), suggesting an increase in autophagosome formation in EC with higher ROS. To further confirm this finding, we transduced MHEC with a construct containing mRFP-EGFP-LC3 that gives off green and red fluorescence signals upon autophagosome formation, but gives off only red signal when autophagosome becomes fused with lysosome to form autolysosome (acidic pH in autolysosome quenches green signal) [25, 40] (Fig. 6A). In accordance with suppression of mTOR signal and increase in LC3A-II, Tet-OFF MHEC demonstrated significant increase in the autophagic events as determined by increased green/red fluorescence compared to Tet-ON MHEC confocal microscopy (Fig. 6B). Quantification of red and green fluorophores showed 1.5±0.18-fold increase in overall autophagy (red plus green signals) in Tet-OFF MHEC compared to Tet-ON MHEC (Fig. 6C). There were, however, no significant changes in the intracellular ratio of red versus green signals in Tet-ON MHEC or in Tet-OFF MHECs (Fig. 6C), and the percentages of colocalization events (overlapping green and red signals) between Tet-ON and Tet-OFF MHEC were similar (Fig. 6D), suggesting that increased autophagy in Tet-OFF MHEC was accompanied by increased autophagosome and autolysosome formation. Together, these data suggest an increased autophagic flux in Tet-OFF MHEC.
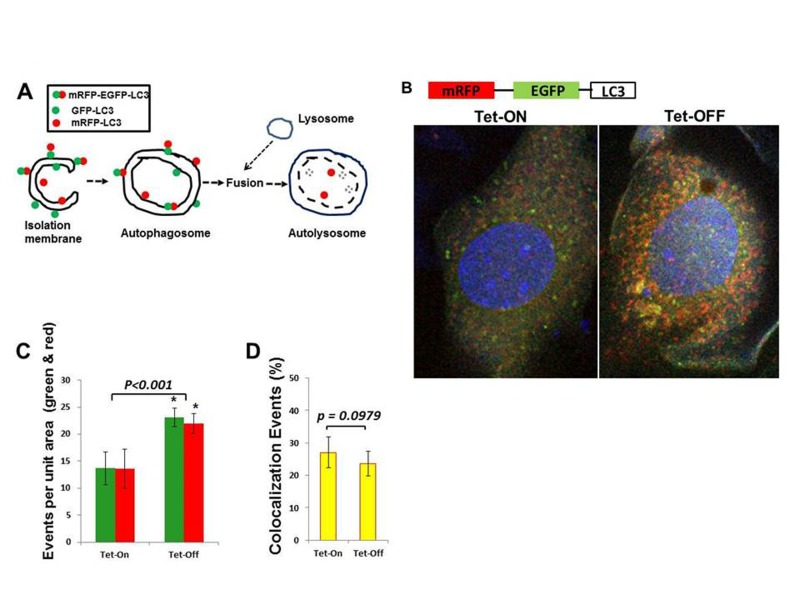
Figure 6. Increased autophagy in Tet-OFF MHEC using adenovirus expressing mRFP-GFP-LC3 (A) Schematic presentation shows that GFP-LC3 (green) and mRFP-LC3 (red) signals are present on the autophagosome, whereas autolysosome contains only mRFP-LC3 (red) signals [40]. (B) Confocal microscopy of Adv-mRFP-GFP-LC3-transduced MHECs. (C) Quantification of red and green fluorophores using NIH ImageJ 1.47b demonstrate >1.5-fold increase in autophagy in Tet-OFF MHEC (n=50 cells). (D) Quantification of colocalization events (yellow) using spatial overlap of red and green was done in Fiji program (ImageJ 1.47h). 1.5-fold increase in overall autophagy in Tet-OFF MHEC (C), but no significant changes in the ratio of intracellular red vs. green in Tet-OFF MHEC (C), and no changes in the colocalization signals between Tet-ON vs. Tet-OFF (D) suggest an effective autophagic flux in Tet-OFF MHEC. *p<0.05.
ROS-induced autophagy plays a protective role in EC survival
To determine whether increased autophagy was due to increased ROS levels, we transfected Tet-OFF MHEC with siRNA-Nox2 (si-Nox2) or pre-treated with NAC (300 μmol/L). Reduction in ROS resulted in inhibition of autophagy in Tet-OFF MHEC (Fig. 7A shows data using si-Nox2), suggesting a role for Nox2-derived ROS in increased autophagy in Tet-OFF MHEC. Next, we wanted to examine whether increased autophagy played a protective for the survival of MHEC with higher ROS levels. Tet-OFF MHEC were treated with chloroquine to inhibit autolysosome formation (chloroquine prevents acidification of lysosomes resulting in inhibition of proteolytic activitiy). Chloroquine-treated Tet-OFF MHEC had significantly higher levels of apoptotic cell death compared to vehicle-treated Tet-OFF MHEC (Fig. 7B), suggesting a protective role for increased autophagy in the survival of ECs with high ROS levels.
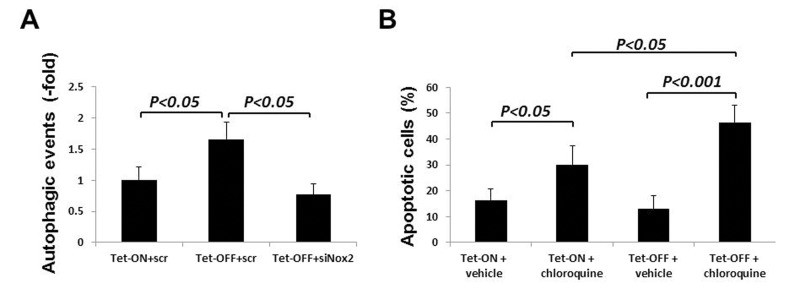
Figure 7. Nox2-induced autophagy plays a protective role in cell survival during oxidative stress in ECs (A) Tet-ON and Tet-OFF MHEC were transduced with Adv-mRFP-GFP-LC3 and transfected with either si-scr or si-Nox2 as indicated. Quantification of red and green fluorophores using NIH ImageJ 1.47b demonstrate significant increase in autophagy in Tet-OFF MHEC (n=50 cells), which was abrogated by knockdown of Nox2. (B) Annexin V-FITC labeling was carried out to determine apoptotic MHEC as described in the Methods. Bar graph shows apoptotic cells as percentage of total viable cells population using three independent experiments. Autophagosome-lysosome fusion blocker chloroquine induced apoptosis in both Tet-ON and Tet-OFF MHEC. However, chloroquine-induced apoptosis was significantly higher in Tet-OFF MHEC.
Discussion
Here, we demonstrate that above-physiological levels of NADPH oxidase-derived ROS in vascular endothelium in vivo induce activation of the pro-survival kinase AMPK, activation of downstream eNOS, and endothelium-dependent coronary vasodilatation. We specifically show that downstream of AMPK, eNOS activation and mTOR inhibition have two distinct positive effects on vascular endothelium during oxidant stress: (i) redox-AMPK-eNOS signaling increases nitric oxide levels to improve endothelium-dependent vasodilatory response in coronary vessels; and (ii) redox-AMPK-mTOR signaling appears to play critical role in endothelial survival by inducing protective autophagy.
These results demonstrate that increased ROS levels improve vascular function. This contradicts the currently believed ‘ROS theory’ and argues against oxidative damage as a probable initial cause of age-associated microvascular pathology. Several recent elegant studies have also challenged the ‘ROS theory’ of aging and organ damage [41-43]. The data presented in this study are obtained using conditional increase (1.8-fold) in NAPDH oxidase-derived ROS in a vascular endothelium-specific manner for four to 12 weeks. There were no changes observed in gross phenotype, fertility or blood pressure in these animals.
The findings suggest existence of an endothelial compensatory mechanism(s) that prevents oxidative damage to the vasculature. Increased synthesis of NO and autophagic flux, as reported in this study, appear to play critical roles in resetting endothelial signaling pathways during oxidative stress. However, further studies are required to determine the effects of prolonged exposure of vascular ECs (beyond 12 weeks) to above-physiological ROS levels that result in ‘hyperfunction’ [41] of AMPK-eNOS (i.e. NO synthesis) and AMPK-mTOR (i.e. autophagy).
Using in vitro cell culture, we and others have previously reported that NADPH oxidase-derived ROS in ECs are involved in the activation of several signaling pathways including PI3K-Akt-eNOS [9, 10, 12, 26, 44]. In the current study, we did not find significant alteration in Akt phosphorylation in the mouse heart ECs (MHEC) with higher redox content, suggesting an apparent disconnect between in vitro and in vivo systems. Interestingly, increased endothelial ROS levels resulted in the activation of the survival kinase AMPK and downstream eNOS. Activation of eNOS by AMPK has been reported earlier in several cardiovascular cells including ECs [45, 46]. AMPK, a kinase known to ‘sense’ cellular metabolic state (AMP vs. ATP), is activated in several cell types following caloric restriction, hypoxia/ischemia, heat shock, and exercise [47, 48]. Two upstream major kinases, LKB1 and CaMKKβ, have been shown to activate AMPK. However, AMPK's role in ECs as a metabolic sensor has not yet been shown and the precise role of AMPK in EC remains incompletely understood. CaMKKβ-mediated activation of AMPK has been shown to occur independent of AMP levels in ECs[49]. Our data support CaMKKβ-mediated AMPK activation in EC with high redox content. To our knowledge, this is the first study demonstrating AMPK activation in ECs in response to increased levels of NADPH oxidase-derived endogenous ROS.
The role of AMPK in the maintenance of vascular tone and vasodilator function remains controversial. Global knockout of AMPK did not affect NO-mediated arterial vasorelaxation in mice[50]. On the contrary, AMPK activator metformin was shown to improve vasodilatation[51, 52]. In addition, several compounds including fenofibrate, rosiglitazone, cilostazol, that activate AMPK were also shown to improve EC-dependent vasodilatation. Resveratrol, a polyphenol known for its cardio-protective and positive survival effects[53], has been reported to increase AMPK activity. The precise mechanism by which AMPK activation brings about these positive changes in vascular health is not known. In the present study, using AMPK-inhibitor compound C and cGMP-inhibitor ODQ, we have shown that ROS-mediated, endothelium-dependent vasodilatation in coronary vessels occurs through activation of AMPK-eNOS-NO axis. These findings suggest an important role for AMPK in the regulation of coronary vasorelaxation during oxidative stress in the vascular endothelium.
Caloric restriction or nutrition deprivation slows down energy-consuming processes and induces autophagy to provide amino acids for the synthesis of essential proteins, resulting in cell survival and inhibition of apoptosis under stressful conditions such as oxidative stress in many cell types. Autophagy is essential for cellular survival, homeostasis, differentiation, and tissue remodeling in physiological and also in pathological conditions. However, depending on the patho-physiological settings, autophagy may play a protective role or contribute to cell damage. Our novel findings that increased ROS levels elicit a ‘caloric restriction’-like response (AMPK-mediated mTOR inhibition and induction of autophagy) in endothelium may have important clinical implication. It is plausible that endothelium deals with higher redox state by slowing down endothelial metabolism and/or by recycling the oxidant-damaged cellular organelles. Further studies are required to address this issue.
Taken together, the findings reported here suggest that NADPH oxidase-derived increase in endothelium-specific ROS induces AMPK-eNOS signaling, NO synthesis and coronary vasodilatation (Fig. 8). Increased ROS also elicit protective mechanisms in coronary endothelium that include AMPK-mTOR-induced autophagy. These findings raise several important questions that need to be addressed in future: (a) what effects would sustained increase in endothelial ROS (beyond 12 weeks) have on coronary vascular function? (b) Do EC-ROS have similar effects on other vascular beds? Preliminary data using aorta demonstrate similar vasodilatory response in Tet-OFF animals (Supplemental Fig. 5). (c) What effects do increased ROS levels have on AMPK-regulated signaling intermediates and transcription factors in vascular endothelium, namely PGC-1α, HIF-1α, and FOXO1? (d) Will redox-AMPK axis modulate endothelial cell growth and proliferation, and thus angiogenesis, in ischemic tissues that often have higher ROS levels, such as IHD? Ongoing studies in our lab are addressing these important questions.
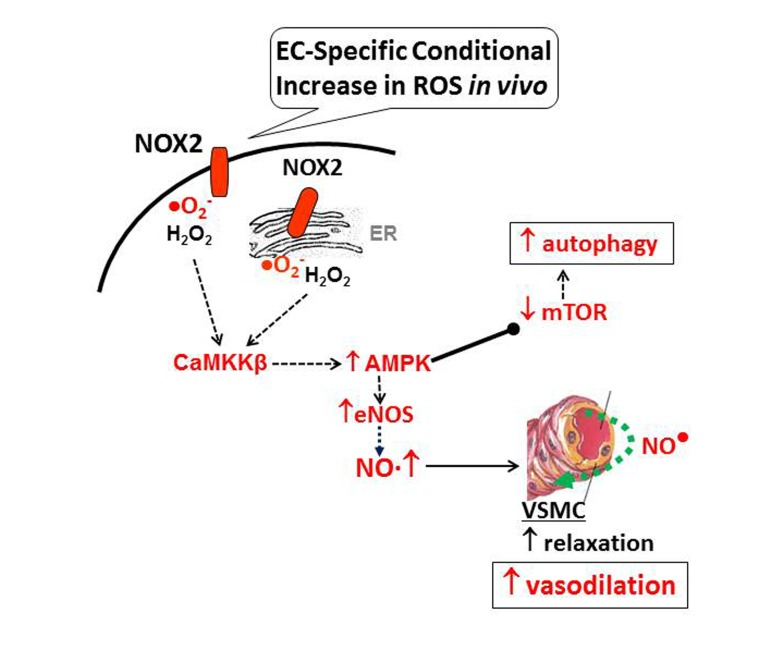
Figure 8. Model for EC-specific ROS-mediated improvement in endothelial function Nox2-induced ROS in vascular endothelium activates CaMKKβ-AMPK, which in turn, activates eNOS to induce NO-mediated vasodilatation and inhibits mTOR resulting in protective autophagy.
Methods
Generation of binary Tet-NOX2: VE-Cadherin-tTA mice
All animal experiments were approved by the Lifespan Institutional Animal Care and Use Committee. The binary tetracycline (Tet)-ON/Tet-OFF system and VE-Cadherin promoter were used to render the transgene inducible and endothelium-specific, respectively [54]. In brief, the HA-tagged NOX2 gene was placed downstream of a Tet (TRE) promoter in one mouse (Tet-NOX2) and the tTA gene was placed under a VE-Cadherin promoter in another mouse (VE-Cad-tTA) (Fig. 1A). Cross-breeding of Tet-Nox2 and VE-Cad-tTA mice were carried out to obtain double transgenic NVF (Tet-Nox2:VE-Cad-tTA) mice. To identify double-positive NVF transgenic mice, PCR was carried out using Platinum® PCR Supermix (Invitrogen). The primer sequences flanking Tet-responsive element (TRE) upstream and Nox2 ORF downstream were used: for sense primer 5'- AGA GAA AAG TGA AAG TCG AGC TCG GT -3', and for antisense primer 5'- TGT GCA GTG CTA TCA TCC AAG C -3'. The primer sequences for VE-Cadherin-tTA were: Sense – 5'- CAG TAG TAG GTG TTT CCC TTT CTT -3', and antisense – 5'- GAC GCC TTA GCC ATT GAG AT -3'. Expression of the transgene (NOX2) was turned-off in double transgenic (NVF) by adding tetracycline (Tet-ON) in the drinking water since in utero. Withdrawal of tetracycline (Tet-OFF) for at least two weeks was required to achieve ~4-fold increase in expression level of Nox2 mRNA and ~1.8-fold increase in ROS levels in ECs (data not shown). For all experiments reported here, tetracycline (Tet) was withdrawn (Tet-OFF) for a minimal period of two weeks to a maximum period of 12 weeks, whereas the sex-matched littermates on Tet were used as control (Tet-ON). HA-tag was used to differentiate between the endogenous gene product and the transgene. All experiments were carried out using age- and sex-matched (male) animals. Two independent lines of the NVF transgenic mice were used in the current study.
Immunofluorescence and immunohistochemistry assays
Immunofluoresence and immunohistochemistry assays were performed as described [55, 56]. Briefly, tissues were harvested and embedded in HistoPrepTM frozen tissue embedding media. Frozen sections were immunostained using anti-CD31 antibody (BD PharmingenTM, 1:250) followed by anti-Rat Alexa 488 secondary antibody. Anti-HA antibody was used to detect expression of the transgene (Novus Biologicals, 1:3000), Microscopy was performed and the Spot Advanced Software was used to capture images in red, green and blue channels. Composite images were also created using Spot Advanced.
Ex Vivo coronary microvessel relaxation studies
After cardiac harvest from 8- to 12-week-old Tet-ON (control) and Tet-OFF Tet-Nox2:VE-Cad-tTA (NVF) mice, coronary arterioles (diameter, 80 to 120 μM; length, 2 mM) were dissected from the surrounding tissue. Microvessel studies [6 mice from each group (Tet-ON and Tet-OFF)] were performed using ex vivo organ bath videomicroscopy, as previously described [10]. Vessels were precontracted using U46619. Where indicated, isolated coronary vessels were pretreated with NO-inhibitor L-NAME (300 μmol/L), ROS inhibitor NAC (400 μmol/L), NO-cGMP inhibitor ODQ (10 μmol/L) and AMPK inhibitor Compound C (80 μmol/L). Endothelial denudation was carried out by advancing a human hair into the lumen of the vessel and gently abrading the luminal surface.
Mouse heart EC isolation and culture
Mouse heart ECs (MHECs) were isolated from the heart specimens of Tet-ON and Tet-OFF animals, as previously described [10]. For each experiment, primary cultures of Tet-ON and Tet-OFF were started simultaneously (a pool of 3 hearts from each group). Tet-ON MHEC were grown in DMEM-based EC culture medium containing tetracycline (2 μg/mL). Results were obtained, in triplicate, using three independent batches of MHEC isolation per group (Tet-ON and Tet-OFF).
Autophagosome/autolysosome formation assay
Tet-ON and Tet-OFF MHEC grown on Lab-Tek Chamber slides were transduced with Ad- mRFP-EGFP-LC3 at 5 MOI (multiplicity of infection) [25, 40]. After 36 hours, MHEC were washed twice with PBS and fixed with 4% paraformaldehyde. DAPI containing mounting medium (Vectashield) was used to identify nuclei. Fluorescence and confocal micrsocopy were used to capture images from three different MHEC preparations for quantification of green, red, and overlapped green-red signals. NIH ImageJ 1.47b was used for quantification of red and green fluorophores (n=50 cells). Fiji program (ImageJ 1.47h) was used to quantify colocalization events (yellow) using spatial overlap of red and green.
Western blots
Cell lysates were prepared from MHEC grown on 0.1% gelatin-coated plates to 80%-90% confluence. Western blot (WB) analyses using MHEC protein lysates were performed as previously described [9, 10]. Anti-phosphorylated (phos) (S473) Akt, anti-phos (S1179) eNOS, anti-phos (T172) AMPK, anti-phos (S2481) mTOR, anti-phos (T389) P70S6K, anti-Nox2, anti-Akt, anti-4E-BP, and anti-LC3A, anti-GAPDH and anti-AMPK antibodies were obtained from Cell Signaling (Beverly, Mass), Abcam (Cambridge, Mass), and Thermoscientific (West Palm Beach, FL). WBs were performed using lysates from three independent experiments and were analyzed using NIH Image J for quantification. * p < 0.05. Representative blots are shown in the Results section.
NADPH oxidase activity and DCF fluorescence assays
Tet-ON and Tet-OFF MHEC were washed with ice-cold PBS, collected by a cell scraper, and Dounce-homogenized in buffer containing 20 mmol/L KH2PO4 (pH 7.0), 1× protease mixture inhibitor (Sigma), 1 mM EGTA, 10 μg/mL aprotinin (Calbiochem), 0.5 μg/mL leupeptin (Sigma), 0.7 μg/mL pepstatin (Sigma), and 0.5 mmol/L phenyl-methylsulfonyl fluoride (Calbiochem). NADPH oxidase activity was measured using a modified assay [57]. Briefly, photon emission from the chromogenic substrate lucigenin (5 μmol/L) as a function of acceptance of electron generated by the NADPH oxidase complex was measured every 15 s for 20 min in a Berthold luminometer as described [9]. Lucigenin activity (light units/min/mg of protein) of control cells (Scram-si-transfected) was arbitrarily set at 100%. Total intracellular levels of ROS were determined by fluorescence-activated cell-sorting (FACS) analyses of the oxidative conversion of cell-permeable 2',7'-dichlorofluorescein diacetate (DCFH-DA; Molecular Probes Inc., Eugene, OR) to fluorescent dichloro-fluorescein as described previously [9]. SOD mimetic MnTBAP (200 μmol/L) and PEG-catalase (250 U/mL) were used to confirm DCF-DA fluorescence was specific to ROS (not lipid peroxides).
Quantitative Real Time PCR
Real time PCR was carried out as described previously[9]. Briefly, RNA was extracted from MHEC using the RNeasy RNA extraction kit (Qiagen, Valencia, CA). Total RNA (100 ng) from each of triplicate samples was converted into cDNA using random primers and SuperscriptIII reverse transcriptase (Invitrogen). Primers were designed using the Primer Express oligo design software (Applied BioSystems, Foster City, CA) and synthesized by Integrated DNA Technologies (Coralville, IA). The level of target gene expression was normalized against the 18 S rRNA expression in each sample, and the data were presented as mRNA copies per 10618 S rRNA copies as described [9].
eNOS activity assay
Tet-ON and Tet-OFF MHEC were lysed in ice-cold homogenization buffer (25-mmol/L Tris·Cl, 1-mmol/L EDTA, and 1-mmol/L EGTA, pH 7.4). eNOS activity was determined using the NOS assay kit (CalBiochem, San Diego, CA) that measures conversion of L-[3H]arginine to L-[3H]citrulline as described previously [10].
Statistical Analyses
All values are presented as mean ± SEM where appropriate. A value of P<0.05 between experimental groups was considered to represent a significant difference (*). Nonlinear regression modeling utilizing the extra sum-of-squares F test to compare slopes (Prism 5. Graph Pad Software) was used for vessel relaxation assays. ANOVA and Tukey's post-hoc test will be used for comparison between groups.
Supplementary Materials
Acknowledgments
We would like to thank Dr. Junichi Sadoshima, UMDNJ for the Ad-mRFP-GFP-LC3. This work was supported by the American Heart Association Grant-in-Aid 10GRNT3640011 to M.R.A., NIH R01 HL46716 to F.W.S., Tide Setubal Foundation/Unibanco to A.L., A.V.D.Z., and Alfaisal University Medical Research Foundation to M.A., A.Y.K.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1. Datla SR and Griendling KK. Reactive oxygen species, NADPH oxidases, and hypertension. Hypertension. 2010; 56: 325 -330. [PubMed] .
- 2. Luo Z, Teerlink T, Griendling K, Aslam S, Welch WJ, Wilcox CS. Angiotensin II and NADPH oxidase increase ADMA in vascular smooth muscle cells. Hypertension. 2010; 56: 498 -504. [PubMed] .
- 3. Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxidants & redox signaling. 2009; 11: 791 -810. [PubMed] .
- 4. Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nature reviews. Drug discovery. 2011; 10: 453 -471. .
- 5. Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. 2003; 361: 2017 -2023. [PubMed] .
- 6. Virtamo J, Rapola JM, Ripatti S, Heinonen OP, Taylor PR, Albanes D, Huttunen JK. Effect of vitamin E and beta carotene on the incidence of primary nonfatal myocardial infarction and fatal coronary heart disease. Arch Intern Med. 1998; 158: 668 -675. [PubMed] .
- 7. Rapola JM, Virtamo J, Ripatti S, Haukka JK, Huttunen JK, Albanes D, Taylor RR, Heinonen PO. Effects of alpha tocopherol and beta carotene supplements on symptoms, progression, and prognosis of angina pectoris. Heart. 1998; 79: 454 -458. [PubMed] .
- 8. Hegele RA. Angiotensin-converting enzyme (ACE) inhibition in the secondary prevention of vascular disease: the Heart Outcomes Prevention Evaluation (HOPE) Trial and its substudies. Curr Atheroscler Rep. 2000; 2: 361 -362. [PubMed] .
- 9. Abid MR and Spokes KC. Shih SCand Aird WC. NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. The Journal of biological chemistry. 2007; 282: 35373 -35385. [PubMed] .
- 10. Feng J, Damrauer SM, Lee M, Sellke FW, Ferran C, Abid MR. Endothelium-dependent coronary vasodilatation requires NADPH oxidase-derived reactive oxygen species. Arteriosclerosis, thrombosis, and vascular biology. 2010; 30: 1703 -1710. .
- 11. Zhang Q, Malik P, Pandey D, Gupta S, Jagnandan D, Belin de Chantemele E, Banfi B, Marrero MB, Rudic RD, Stepp DW, Fulton DJ. Paradoxical activation of endothelial nitric oxide synthase by NADPH oxidase. Arteriosclerosis, thrombosis, and vascular biology. 2008; 28: 1627 -1633. .
- 12. Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circulation research. 2002; 91: 1160 -1167. [PubMed] .
- 13. Sawada N, Salomone S, Kim HH, Kwiatkowski DJ, Liao JK. Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circulation research. 2008; 103: 360 -368. [PubMed] .
- 14. Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K, Keaney JF Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. 2011; 124: 731 -740. [PubMed] .
- 15. Cai H, Li Z, SDikalov S, Holland SM, Hwang J, Jo H, Dudley SC Jr, Harrison DG. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. The Journal of biological chemistry. 2002; 277: 48311 -48317. [PubMed] .
- 16. Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arteriosclerosis, thrombosis, and vascular biology. 2011; 31: 1368 -1376. .
- 17. Krotz F, Engelbrecht B, Buerkle MA, Bassermann F, Bridell H, Gloe T, JDuyster J, Pohl U, Sohn HY. The tyrosine phosphatase, SHP-1, is a negative regulator of endothelial superoxide formation. Journal of the American College of Cardiology. 2005; 45: 1700 -1706. [PubMed] .
- 18. Lee M, Choy WC, Abid MR. Direct sensing of endothelial oxidants by vascular endothelial growth factor receptor-2 and c-Src. PloS one. 2011; 6: e28454 [PubMed] .
- 19. Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Molecular and cellular biology. 2011; 31: 3531 -3545. [PubMed] .
- 20. Mackenzie RM, Salt IP, Miller WH, Logan A, Ibrahim HA, Degasperi A, Dymott JA, Hamilton CA, Murphy MP, Delles C, Dominiczak AF. Mitochondrial reactive oxygen species enhance AMP-activated protein kinase activation in the endothelium of patients with coronary artery disease and diabetes. Clinical science. 2013; 124: 403 -411. [PubMed] .
- 21. Wang Q, Liang B, Shirwany NA, Zou MH. 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PloS one. 2011; 6: e17234 [PubMed] .
- 22. Lee M, Hwang JT, Lee HJ, Jung SN, Kang I, Chi SG, Kim SS, Ha J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. The Journal of biological chemistry. 2003; 278: 39653 -39661. [PubMed] .
- 23. Dixit M, Bess E, Fisslthaler B, Hartel FV, Noll T, Busse R, IFleming I. Shear stress-induced activation of the AMP-activated protein kinase regulates FoxO1a and angiopoietin-2 in endothelial cells. Cardiovascular research. 2008; 77: 160 -168. [PubMed] .
- 24. Cardaci S, Filomeni G, Ciriolo MR. Redox implications of AMPK-mediated signal transduction beyond energetic clues. Journal of cell science. 2012; 125: 2115 -2125. [PubMed] .
- 25. Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007; 3: 452 -460. [PubMed] .
- 26. Abid MR, Tsai JC, Spokes KC, Deshpande SS, Irani K, Aird WC. Vascular endothelial growth factor induces manganese-superoxide dismutase expression in endothelial cells by a Rac1-regulated NADPH oxidase-dependent mechanism. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001; 15: 2548 -2550. [PubMed] .
- 27. Bayraktutan U, Blayney L, Shah AM. Molecular characterization and localization of the NAD(P)H oxidase components gp91-phox and p22-phox in endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. 2000; 20: 1903 -1911. .
- 28. Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS, Lambeth JD. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99: 715 -720. [PubMed] .
- 29. Bengtsson M SH, Gulluyan LM, Dusting GJ, Drummond GR. Novel isoforms of NADPH oxidase in vascular physiology and pathophysiology. Clinical and experimental pharmacology & physiology. 2003; 30: 849 -854. [PubMed] .
- 30. Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J, Wakisaka M, Ibayashi S, Utsumi H, Iida M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004; 109: 227 -233. [PubMed] .
- 31. BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, Gorlach A. NOX5 variants are functionally active in endothelial cells. Free radical biology & medicine. 2007; 42: 446 -459. [PubMed] .
- 32. Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxidants & redox signaling. 2005; 7: 308 -317. [PubMed] .
- 33. Thomas SR, Chen K, Keaney JF Jr. Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. The Journal of biological chemistry. 2002; 277: 6017 -6024. [PubMed] .
- 34. Cai H, Davis ME, Drummond GR, Harrison DG. Induction of endothelial NO synthase by hydrogen peroxide via a Ca(2+)/calmodulin-dependent protein kinase II/janus kinase 2-dependent pathway. Arteriosclerosis, thrombosis, and vascular biology. 2001; 21: 1571 -1576. .
- 35. Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circulation research. 2012; 110: 1217 -1225. [PubMed] .
- 36. Bendall JK, Rinze R, Adlam D, Tatham AL, de Bono J, Wilson N, Volpi E, Channon KM. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial-targeted Nox2 transgenic mice. Circulation research. 2007; 100: 1016 -1025. [PubMed] .
- 37. Zhang Q, Malik P, Pandey D, Gupta S, Jagnandan D, Belin de Chantemele E, Banfi B, Marrero MB, Rudic RD, Stepp DW, Fulton DJ. Paradoxical activation of endothelial nitric oxide synthase by NADPH oxidase. Arterioscler Thromb Vasc Biol. 2008; 28: 1627 -1633. [PubMed] .
- 38. Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Current opinion in cell biology. 2005; 17: 596 -603. [PubMed] .
- 39. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circulation research. 2007; 100: 914 -922. [PubMed] .
- 40. Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxidants & redox signaling. 2011; 14: 2179 -2190. [PubMed] .
- 41. Gems D and Guardia YD. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxidants & redox signaling. 2012; .
- 42. Blagosklonny MV. Aging: ROS or TOR. Cell cycle. 2008; 7: 3344 -3354. [PubMed] .
- 43. Blagosklonny MV. Hormesis does not make sense except in the light of TOR-driven aging. Aging. 2011; 3: 1051 -62. [PubMed] .
- 44. Sessa W.C. eNOS at a glance. Journal of cell science. 2004; 117: 2427 -2429. [PubMed] .
- 45. Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS letters. 1999; 443: 285 -289. [PubMed] .
- 46. Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. The Journal of biological chemistry. 2003; 278: 31629 -31639. [PubMed] .
- 47. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nature reviews. Molecular cell biology. 2007; 8: 774 -785. .
- 48. Fisslthaler B and Fleming I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circulation research. 2009; 105: 114 -127. [PubMed] .
- 49. Stahmann N, Woods A, Carling D, Heller R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Molecular and cellular biology. 2006; 26: 5933 -5945. [PubMed] .
- 50. Goirand F, Solar M, Athea Y, Viollet B, Mateo P, Fortin D, Leclerc J, Hoerter J, Ventura-Clapier R, Garnier A. Activation of AMP kinase alpha1 subunit induces aortic vasorelaxation in mice. The Journal of physiology. 2007; 581: 1163 -1671. [PubMed] .
- 51. Katakam PV, Ujhelyi MR, Hoenig M, Miller AW. Metformin improves vascular function in insulin-resistant rats. Hypertension. 2000; 35: 108 -112. [PubMed] .
- 52. Sartoretto JL, Melo GA, Carvalho MH, Nigro D, Passaglia RT, Scavone C, Cuman RK, Fortes ZB. Metformin treatment restores the altered microvascular reactivity in neonatal streptozotocin-induced diabetic rats increasing NOS activity, but not NOS expression. Life sciences. 2005; 77: 2676 -2689. [PubMed] .
- 53. Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewi Ks, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006; 444: 337 -342. [PubMed] .
- 54. Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I, Nagy JA, Lin MI, Walsh K, Dvorak AM, Briscoe DM, Neeman M, Sessa WC, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006; 10: 159 -170. [PubMed] .
- 55. Abid MR, Yano K, Guo S, Patel VI, Shrikhande G, Spokes KC, Ferran C, Aird WC. Forkhead transcription factors inhibit vascular smooth muscle cell proliferation and neointimal hyperplasia. J Biol Chem. 2005; 280: 29864 -29873. [PubMed] .
- 56. Mukai Y, Rikitake Y, Shiojima I, Wolfrum S, Satoh M, Takeshita K, Hiroi Y, Salomone S, Kim HH, Benjamin LE, Walsh K, Liao JK. Decreased vascular lesion formation in mice with inducible endothelial-specific expression of protein kinase Akt. J Clin Invest. 2006; 116: 334 -343. [PubMed] .
- 57. Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circulation research. 1994; 74: 1141 -1148. [PubMed] .
- 58. Abid MR, Guo S, Minami T, Spokes KC, Ueki K, Skurk C, Walsh K, Aird WC. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. 2004; 24: 294 -300. .