




Introduction
Sarcopenia is an advanced age-related loss of skeletal muscle and function, which reduces the amount of metabolically active tissue, thus, increasing the risk for metabolic diseases [1, 2], and negatively impacts mobility, limiting the independence living and quality of life of elderly individuals [3, 4]. The course of sarcopenia and age-related diseases that are associated with sarcopenia [1, 5] involve complex processes that are controlled by both extrinsic and intrinsic factors, many of which converge on a decline in the ability of muscle stem cells (satellite cells) to replace and repair damaged muscle fibers in old hosts [6-8].
Although the mechanisms that initiate of sarcopenia are largely unknown, an increased production and accumulation of reactive oxygen species (ROS) has been proposed to underlie the pathogenicity of sarcopenia [9, 10]. Oxidative stress occurs when the production of oxidants exceeds the capacity of the cells to eliminate or buffer oxidizing reactions to proteins, DNA and lipids. The elevated levels of oxidized molecules may contribute to the progression of sarcopenia by controlling many cellular pathways including redox-sensitive signaling pathways [11]. The mitochondria are both ROS producers and are adversely affected by excessive ROS levels. For example, a comparative electron-microscopic study of the ultrastructure of mitochondria in skeletal muscles of young (low basal ROS levels) and old rats (high basal levels of ROS) revealed age-dependent changes in both the general organization of the mitochondrial reticulum and the ultrastructure of mitochondria [12], which presumably contributes to the age-associated dysfunction of this organelle. Furthermore, early treatment of aged mice with the mitochondrial antioxidant SkQ1 was shown to prevent the development of age-dependent destructive pathological changes in mitochondria [12].
One group of factors sensitive to changes in the cellular redox pathways is the poly(ADP-ribose) polymerases (PARPs). ROS have a robust ability to induce the poly (ADP-ribosyl)ation (PARylation) of many proteins, which regulate the cell cycle, growth, and survival, thereby positioning PARylation as an important biochemical marker of oxidative stress [13].
PARylation is one of the post-translational modifications of proteins that are regulated by the PARP family of enzymes in most eukaryotic organisms [14] PARPs catalyze the covalent transfer of mono or poly(ADP) units from nicotinamide adenine dinucleotide (NAD+) to glutamate or aspartate residues within target proteins, resulting in the synthesis of a large chain of branched ADP-ribose polymers [15]. This post-translational modification either alters the functional properties of PAR-binding proteins or allows the proteins to be degraded by poly (ADP-ribose) glycohydrolase [16]. Surprisingly, many of the putative PAR-binding proteins regulate a wide range of cellular functions including cell survival [13]. PARP-1 is the most extensively studied PARP family protein that requires a source of nuclear NAD+ for its function [17, 18]. Although basal activation of PARP-1 is necessary to maintain normal cell homeostasis, over activation of PARP-1 by ROS species such as superoxide (O2−) and hydrogen peroxide (H2O2) increases protein PARylation and depletes intracellular NAD+ levels leading to cell death [19]. This effect suggests that tight regulation of PARP-1 activity is important for cell survival.
Many lines of evidence have shown that caloric restriction is an effective intervention to slow the aging process in most organisms, and thereby delaying the onset of age-related disease and functional decline [20, 21] Although a wide range of signaling pathways regulates the effects of caloric restriction on aging, Silent information regulator 1 (SIRT-1) has emerged as a promising target from these pathways [22-26]. SIRT-1 has been shown to inhibit the differentiation of mouse C2C12 myoblasts and reduce the expression of myogenin, which is an important regulator for the myogenic specification and differentiation of activated satellite cells [27, 28]. Furthermore, SIRT-1 has been shown to directly induce proliferation of satellite cells [29]. These findings suggest that SIRT-1 may have an important role in prolonging or enhancing proliferation of satellite cells. However, while satellite cell function is reduced with aging, and thus, lower SIRT-1 protein levels might be expected in muscles of old animals, it is interesting to note that increased levels of SIRT-1 have been reported in satellite cells isolated from old rats [30], although the significance of this is not clear. One possibility to explain this complex role of SIRT-1 in skeletal muscle is that SIRT-1 activity and not the abundance of this protein may be more important for determining the downstream function in sarcopenic muscle as it is in muscles of young animals [31]. Thus, understanding the pathways that regulate the activity of SIRT-1 is critical, especially in aging muscles.
SIRT-1 is an NAD+-dependent protein deacetylase, modulation of nuclear NAD+ levels can alter the activity of SIRT-1 in skeletal muscle [32, 33], which is presumably independent of any changes in protein levels occurring with aging. However, high cellular NAM and/or NADH levels inhibit SIRT-1 activity [34], suggesting that increasing cellular NAD+ levels would be an effective way to activate the SIRT-1 pathway in aging muscle. SIRT-1 and PARP-1 regulate many common pathways, including oxidative stress responses and cell survival. Furthermore, SIRT-1 and PARP-1 compete for the same NAD+ pool. As a result, if PARP-1 utilizes NAD+ at a high rate to increase its activity, we would anticipate that SIRT-1 will have less NAD+ available to it, which in turn should suppress the activity of SIRT-1. [32, 35]. This idea is consistent with observations showing that activation of PARP-1 upon cellular stress depletes intracellular NAD+ stores and subsequently releases high levels of NAM, which in turn, significantly inhibits SIRT-1 activity [32, 36, 37]. These findings suggest that these two proteins might be able to counterbalance each other's activity to regulate cell survival. However, the mechanism by which SIRT-1 inhibits the activity of PARP-1 and protects skeletal muscles from ROS-induced decline in muscle performance is unknown. We show here that exercise increases the activity of SIRT-1, which physically binds to and inhibits PARP-1 activity via a deacetylation-dependent mechanism in skeletal muscles from young mice. This protective role of SIRT-1 dramatically declined in skeletal muscles from aged mice as a result of PARP-1 over-activation. Pharmacological inhibition of PARP-1 restored the effect of SIRT-1, and as a result increased skeletal muscle performance in aged mice. Moreover, the enhanced-activity of PARP-1 in skeletal muscles from aged mice is due to an elevated level of acetylation of PARP-1 by General control of amino acid synthesis protein 5-like 2 (GCN5). These results suggest that activation of SIRT-1 and/or inhibition of PARP-1 could be a therapeutic strategy for the treatment of the age-associated decline in muscle performance and potentially counterbalance the functional decay in sarcopenia.
Results
SIRT-1 protects skeletal muscle in young mice from PARP-1 by deacetylation-dependent mechanism
To study the interplay between SIRT-1 and PARP-1 in vivo, we induced oxidative stress in skeletal muscles of young mice using electrically evoked isometric-exercise, which has been shown to generate ROS in skeletal muscle [9, 38]. Electrically evoked exercise modestly increased PARP-1 activity in skeletal muscles, as evidenced by global protein PARylation (Fig. 1A) without altering the mRNA and protein levels of PARP-1 (Fig. 1B). In contrast, exercise significantly increased SIRT-1 mRNA and protein levels (Fig. 1C) as well as SIRT-1 activity, as evidenced by PGC-1α hypo-acetylation (Fig. 1D). The increased activation of PGC-1α resulted in up-regulation of the genes necessary for mitochondrial biogenesis and oxidative metabolism in exercised-muscle (Fig. 1E). The increased mtDNA content was confirmative of an elevation in mitochondrial biogenesis (Fig. 1F). PARP-1 activity can be modulated by its acetylation level [37, 39], and therefore, we explored the acetylation of PARP-1, as a potential mechanism that would explain the decline in PARP-1 activity in skeletal muscle from young mice. IP assays using cell lysates from exercised and non-exercised skeletal muscles demonstrated that exercise decreased the acetylation of PARP-1 (Fig. 1G), suggesting that the reduced activity of PARP-1 in exercised skeletal muscle might be due to hypo-acetylation of PARP-1. Because SIRT-1 can deacetylate non-histone proteins, and given that exercise increased the activity of SIRT-1 (Fig. 1C and D), we sought to determine if SIRT-1 could deacetylate PARP-1. Data from IP assays showed an increased association between SIRT-1 and PARP-1 proteins in exercised muscles as compared to intra-animal non-exercised muscles (Fig. 1H). Earlier studies have shown that PCAF and p300/CBP are the acetyltransferases of PARP-1 [37, 39]. Therefore, we sought to determine whether the decreased acetylation of PARP-1 was due to a reduced association between PARP-1 and PCAF or p300/CBP. Surprisingly, we could not detect any significant association between PARP-1 and p300/CBP or PCAF (data not shown). However, we did observe a substantial interaction between PARP-1 and GCN5 in which electrically-evoked exercise significantly decreased the association between GCN5 and PARP-1 proteins (Fig. 1I). Concomitant with the decreased association of GCN5 with PARP-1, the level of GCN5 protein was also significantly lower in exercised-skeletal muscle (Fig. 1J). These results indicate that the potential mechanism whereby SIRT-1 protects skeletal muscle in young mice from the elevated PARP-1 activity that is seen in old age, is via increased deacetylation-dependent inactivation and/or reduced GCN5 levels in response to exercise.
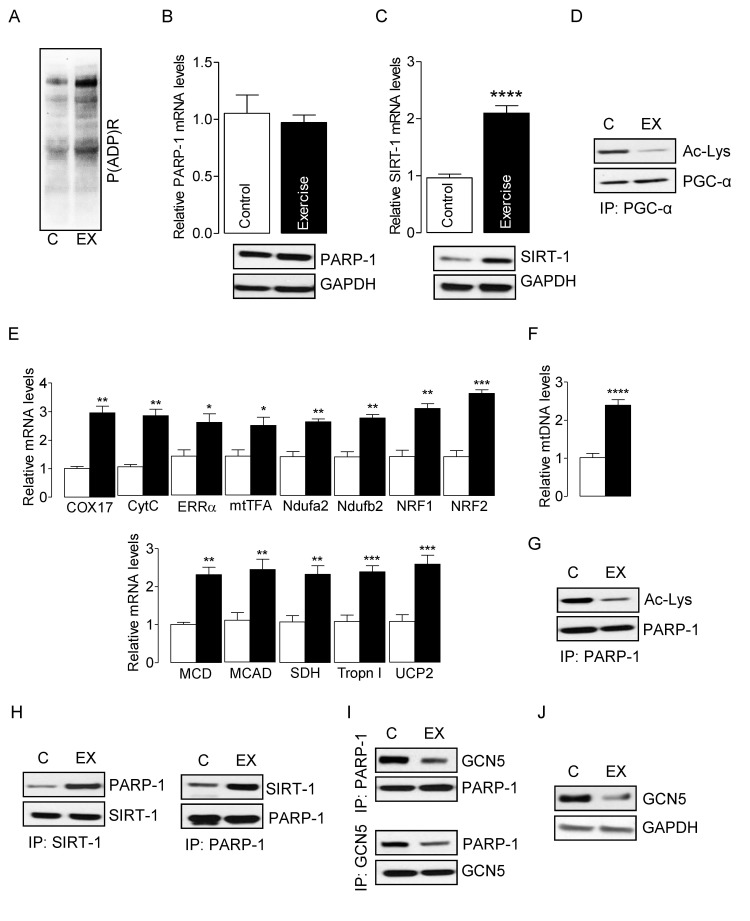
Figure 1. SIRT-1 deactivates exercise-induced PARP-1 in skeletal muscle from young mice Total RNA and cell lysates were isolated from the control or exercised gastrocnemius muscle. (A) Global cellular protein PARylation was determined in total cell lysates by immunoblots. PARP-1 (B) and SIRT-1 (C) mRNA (top) and protein (bottom) levels were determined in total muscle mRNA and cell lysate, respectively. GAPDH was used as a loading control. (D) PGC-1α acetylation levels were estimated by immunoblotting after IP. (E) mRNA expression of the indicated genes in the total RNA was examined by qPCR. (F) mtDNA was evaluated in total muscle genomic DNA by qPCR. (G) PARP-1 acetylation levels were estimated by immunoblotting after IP. (H) SIRT-1 and PARP-1 or (I) GCN5 and PARP-1 binding assays were estimated by immunoblotting after IP. (J) GCN5 protein levels were determined in total cell lysates by immunoblots. The blots are representative of three independent experiments. The data are presented as mean ± SEM (n = 3). White and black bars indicate non-exercised and exercised gastrocnemius muscle, respectively. COX17, cyclooxygenase 17; CytC, Cytochrome C; ERR-α, estrogen-related receptor α; mtTFA, mitochondrial transcription factor A; Ndufa2, NADH dehydrogenase [ubiquinone] iron-sulfur protein a 2; NRF1,nuclear respiratory factor 1; MCD, medium-chain acyl-CoA dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; SDH, succinate dehydrogenase; Tropn I, troponin I; UCP2, uncoupling protein 2; immunoprecipitation (IP).
Dysregulation of SIRT-1 increases PARP-1 activity and reduces skeletal muscle performance in aged mice
In skeletal muscles of aged mice, in which the basal oxidant level is already high, exercise has the ability to further increase oxidant production by as much as 80% [40]. As SIRT-1 inactivates PARP-1 and protects skeletal muscles in young mice, we sought to determine whether SIRT-1 could exert a similar effect in skeletal muscle from aged mice. Electrically evoked exercise robustly increased PARP-1 activity, as indicated by elevated global protein PARylation (Fig. 2A) and also reduced the intracellular NAD+ content (Fig. 2B) without changing PARP-1 mRNA and protein levels (Fig. 2C). Electrically-evoked exercise did not alter SIRT-1 mRNA and protein levels (Fig. 2D), or PGC-1α acetylation status, a result that was in contrast with the data from young mice (Fig. 2E). IP assays showed that the association between SIRT-1 and PARP-1 proteins in skeletal muscles from aged mice was greatly lower in response to exercise, which is in contrast to what we had observed for young mice (Fig. 2F). In agreement with these results, the acetylation level of PARP-1 was increased (Fig. 2G) due to an increased association of GCN5 with PARP-1 (Fig. 2H) in skeletal muscle from aged mice. To establish the role of PARP-1 acetylation by GCN5, we measured the mRNA and protein levels of GCN5. As illustrated in Fig. 2I, there were no changes in the levels of GCN5 mRNA and protein. As the acetylation status of PGC-1α was unchanged, it was not surprising to find that the genes that were necessary for mitochondrial biogenesis and oxidative metabolism in exercised-muscle were significantly lower in muscles from aged mice (Fig. 2J). To explore the dysregulation of SIRT-1 and over-activation of PARP-1 could change the muscle performance; we determined maximal isometric forces from the plantar flexor muscle of aged mice. As illustrated in Figure 2K, aged mice had an increased in vivo plantarflexor muscle fatigue as compared to young mice. This was shown by a shift to the right in the fatigue index after 30 contractions as compared to young mice. These results suggest that dysregulation of SIRT-1 in aged mice decreased mitochondrial biogenesis and oxide-tive metabolism, and increased PARP-1 activity, which in turn, increased muscle fatigue in response to exercise.
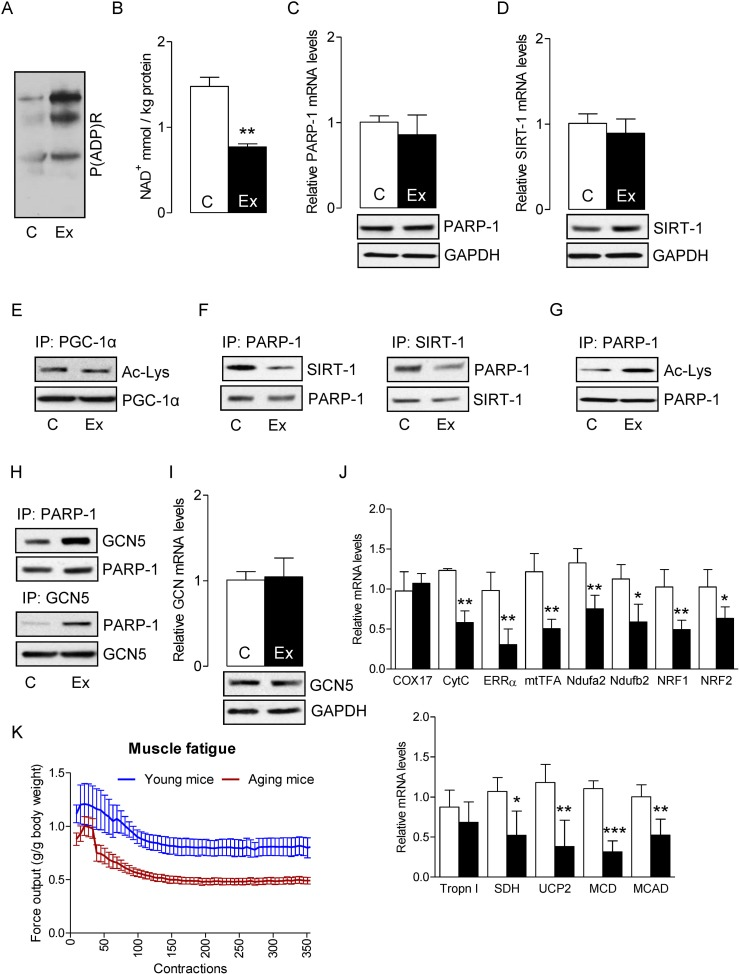
Figure 2. SIRT-1 dysregulation in aged mice increases skeletal muscle fatigue Total RNA and cell lysates were isolated from the control or exercised gastrocnemius muscles. (A) Global cellular protein PARylation was determined in total cell lysate by immunoblots. (B) NAD+ levels in skeletal muscle were determined in control and exercised muscles. PARP-1 (C) and SIRT-1 (D) mRNA (top) and protein (bottom) levels were determined in total muscle mRNA and cell lysates, respectively. GAPDH was used as a loading control. (E) PGC-1α acetylation levels were estimated by immunoblotting after IP. (F) SIRT-1 and PARP-1 binding assays, PARP-1 acetylation levels (G) or PARP-1 and GCN5 binding assays (H) were estimated by immunoblotting after IP. (I) GCN5 mRNA (top) and protein (bottom) levels were determined in total muscle mRNA and cell lysate, respectively. (J) mRNA expression of the indicated genes in the total RNA was examined by qPCR. (K) Maximal evoked isometric forces from 350 contractions are shown for the plantar flexor muscles in young and aged mice. All force measurements were normalized to body weight (g). The blots are representative of three independent experiments. The data are presented as mean ± SEM (n = 3). White and black bars indicate non-exercised and exercised gastrocnemius muscles, respectively. COX17, cyclooxygenase 17; CytC, Cytochrome C; ERR-α, estrogen-related receptor α; mtTFA, mitochondrial transcription factor A; Ndufa2, NADH dehydrogenase [ubiquinone] iron-sulfur protein a 2; NRF1,nuclear respiratory factor 1; MCD, medium-chain acyl-CoA dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; SDH, succinate dehydrogenase; Tropn I, troponin I; UCP2, uncoupling protein 2; immunoprecipitation (IP).
Administration of the PARP-1 inhibitor PJ34 in aged mice increases SIRT-1- mediated mitochon-drial biogenesis and skeletal muscle performance
We hypothesized that inhibition of PARP-1 by the PARP-1 inhibitor PJ34 would increase the activity of SIRT-1 by elevating intracellular NAD+. To test this hypothesis, we injected PJ34 intraperitoneally in aged mice (10 mg/kg bw) 24 h prior to exercise. In agreement with our hypothesis, inhibition of PARP-1 by PJ34 reduced the exercise-induced activation of PARP-1 (Fig. 3A) and this was followed by increasing the intracellular NAD+ content (Fig. 3B). Given the impact of PJ34 on the modulation of NAD+ levels, it was not surprising to observe that mice injected with the PARP-1 inhibitor displayed higher SIRT-1 activity, as demonstrated by reduced PGC-1α acetylation (Fig. 3C). However, inhibition of PARP-1 by PJ34 did not alter PARP-1 and SIRT-1 levels (Fig. 3D and E). The increased SIRT-1 activity coincided with a decreased acetylation of PARP-1 (Fig. 3F) that was due to a strong interaction between SIRT-1 and PARP-1, (Fig. 3G) and a weak interaction between GCN5 and PARP-1 (Fig. 3H). The increased activation of PGC-1α resulted in the up-regulation of genes that are necessary for mitochondrial biogenesis and oxidative metabolism in exercised-muscle (Fig. 3I). The increased content of mtDNA further confirmed the mitochondrial biogenesis (Fig. 3J). The initial rate of fatigue for the plantarflexors was reduced in PJ34-injected mice when compare to PBS injected mice (Fig. 3K). The initial force production in the PJ34 treated animals was ~ 11.3% greater than the PBS treated animals. Force typically increases for the first ~ 30 contractions in both animal groups, likely as a result of having enhanced calcium release, then force declined thereafter as fatigue occurred. However, the average percent of force decline (i.e. fatigue index= [contraction 1-contraction 50/contraction 1x100]) was greater in control (−15.4 ± 7.1%) than in PJ34 (−0.2 ± 8.4%) treated animals after the first 50 contractions, suggesting that PJ34 reduced the initial rate of fatigue thereby delaying the onset of fatigue. Similarly, the average fatigue index after 60 contractions was greater in PBS control as compared with PJ34 treated animals (−19.1 ±9.4% vs. −9.5 ± 6.9%). However, there were no differences between the rate of fatigue after 70 (−25.7 ±6.9% vs. −19.9 ± 7.6%), 80 (−18.8 ±6.7% vs. −14.4 ± 8.3%), 100, (−31.8 ±7.5% vs. −23.3 ± 7.1%), 125 (−20.4 ±4.1% vs. −27.5 ± 12.9%), or 150 (−39.2 ±8.1% vs. −29.8 ± 13.1%) contractions in control and PJ34 treated animals, respectively. Thus, while PJ34 appeared to delay fatigue by sustaining force production over the early series of contractions (<60 contractions), it did not improve the rate of fatigue per se after 60 contractions. Nevertheless, PJ34 did permit a greater total work (area under the force x time curve) to be performed over the course of the experiment such that PJ34 supplemented animals performed 34% more work at the beginning of the contractions, and this leveled off to ~20.4% more work by the 150th contraction and stayed at this level throughout the remainder of the 360 contractions. Together these results suggest that the inhibition of PARP-1 may be an effective way to rescue skeletal muscles from ROS/PARP-1-induced decline in muscle performance (e.g., delay the onset of fatigue and increase the total sustained work) in sarcopenia.
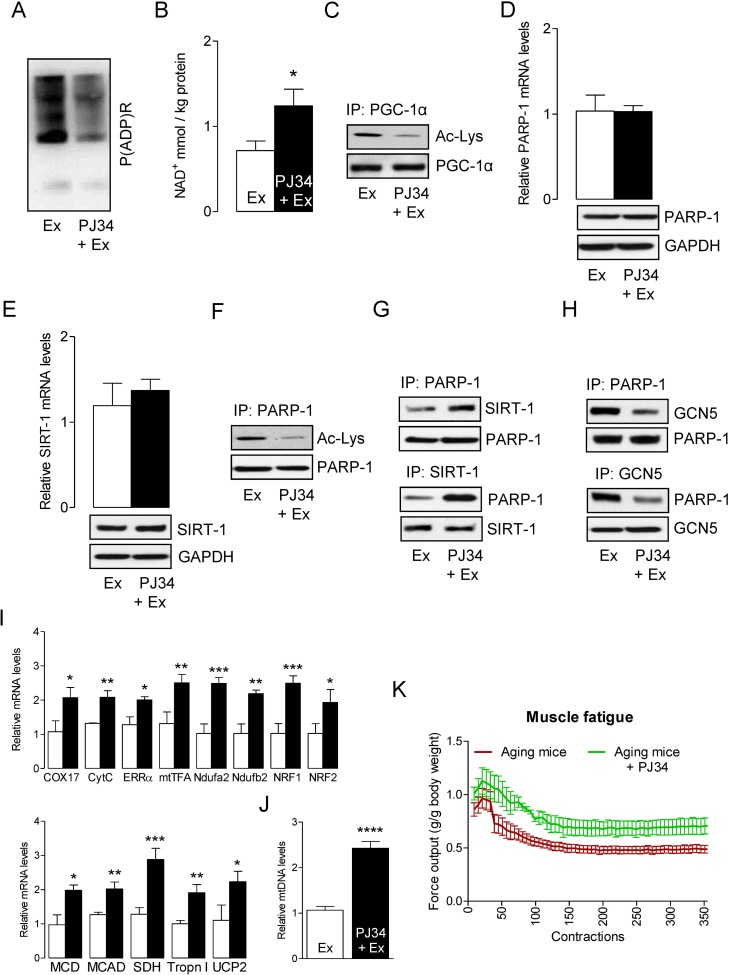
Figure 3. PARP-1 inhibition increases SIRT-1 activity and improves skeletal muscle fatigue Total RNA and cell lysates were isolated from the skeletal muscle of mice that were exercised and PBS injected (Ex) or exercised and PJ34 injected (Ex + PJ34). (A) Global cellular protein PARylation was determined in total cell lysates by immunoblots. (B) NAD+ levels were determined in in skeletal muscle of Ex or Ex + PJ34 mice. (C) PGC-1α acetylation levels were estimated by immunoblotting after IP. PARP-1 (D) and SIRT-1 (E) mRNA (top) and protein (bottom) levels were determined in total muscle mRNA and cell lysates, respectively. GAPDH was used as a loading control. PARP-1 acetylation levels (F), SIRT-1 and PARP-1 binding assays (G), or PARP-1 and GCN5 binding assays (H) were estimated by immunoblotting after IP. (I) mRNA expression of the indicated genes in the total RNA was examined by qPCR. (J) mtDNA was evaluated in total muscle genomic DNA by qPCR. (K) Maximal isometric forces from the plantar flexor muscles are presented for Ex or Ex + PJ34 mice. All force measurements were normalized to body weight (g). The blots are representative of three independent experiments. The data are presented as mean ± SEM (n = 3). White and black bars indicate Ex and Ex + PJ34 mice, respectively. COX17, cyclooxygenase 17; CytC, Cytochrome C; ERR-α, estrogen-related receptor α; mtTFA, mitochondrial transcription factor A; Ndufa2, NADH dehydrogenase [ubiquinone] iron-sulfur protein a 2; NRF1,nuclear respiratory factor 1; MCD, medium-chain acyl-CoA dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; SDH, succinate dehydrogenase; Tropn I, troponin I; UCP2, uncoupling protein 2. IP, immunoprecipitation.
PARP-1 inhibition prevents ROS-induced myotube death via the SIRT-1 pathway
Excessive ROS induces necrotic- and apoptotic-mediated cell death [41]. To determine the role of PARP-1 on the survival of myotubes, we incubated myotubes with H2O2 (300 μM for4 h), because H2O2 is the most stable form of ROS and an established inducer of PARP-1 activity [42]. Myotubes treated with H2O2 robustly increased PARP-1 activity, as evidenced by higher global protein PARylation (Fig. 4A) without changing PARP-1 mRNA and protein levels (Fig. 4B). H2O2-treatment also significantly reduced the survival of myotubes in a time-dependent manner (Fig. 4C). However, incubation of myotubes with the PARP-1 inhibitor PJ34 (1 μM for 24 h) before H2O2 treatment substantially decreased PARP-1 activity (Fig. 4D) and increased the survival of myotubes (Fig. 4E). PARP-1 is the predominant PARP isoform in most tissues and accounts for about 90% of total cellular PARP activity [43]. Therefore, we used RNAi to suppress the level of endogenous PARP-1 in the myotubes (Fig. 4F). As expected, knockdown of PARP-1 decreased both the H2O2-induced PARP-1 activation (Fig. 4G) and myotube death (Fig. 4H), confirming the role of PARP-1 in ROS-induced myotube death. Next, we studied the impact of over activity of PARP-1 on SIRT-1 function. Myotubes treated with H2O2 did not exhibit changes in SIRT-1 mRNA and protein levels (Fig. 4I). However, H2O2-treatment did cause a substantial reduction in SIRT-1 activity, as shown by PGC-1α hyper-acetylation, and pre-incubation of the myotubes with PJ34 or PARP-1-targeting siRNAs blunted this effect (Fig. 4J). Finally, we determined if over-activation of SIRT-1 could rescue myotubes from H2O2/PARP-1-induced cell death. Treatment of myotubes with the SIRT-1 activator resveratrol (25 μM for 24 h) significantly increased SIRT-1 activity (Fig. 4K) and markedly reduced H2O2-mediated PARP-1 activity (Fig. 4L) and myotube death (Fig. 4M). These results indicate that SIRT-1 promotes myotube survival and prevents PARP-1-induced myotube death.
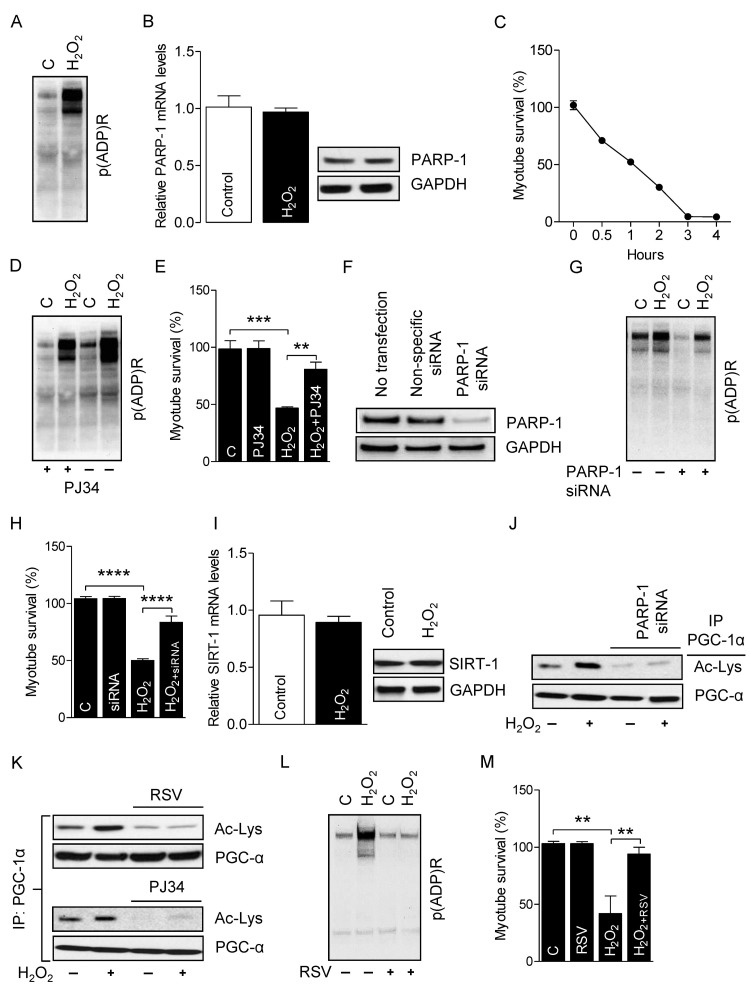
Figure 4. PARP-1 inhibition prevents the H2O2-induced myotube death Total RNA and cell lysates were isolated from myotubes either treated or not treated with H2O2. (A) Global cellular protein PARylation was determined in the total cell lysate by immunoblots. (B) PARP-1 mRNA and protein levels were determined in total mRNA and cell lysates, respectively. GAPDH was used as a loading control. (C) Myotube survival (%) in the presence or absence of H2O2 was determined by MTT assay at the indicated time-points. (D) Global cellular protein PARylation was determined in total cell lysates from either H2O2-treated or non-treated myotubes either in the presence or absence of PJ34 by immunoblots. (E) Myotube survival (%) was determined by the MTT assay in myotubes treated with the conditions similar to ‘D’. (F) Myotubes were transfected with non-specific or PARP-1-targeting siRNAs. PARP-1 protein abundance were analyzed 48 h after transfection by immunoblots. (G) Global cellular protein PARylation was determined in total cell lysates from either H2O2-treated or non-treated myotubes either in the presence or absence of PARP-1 by immunoblots. (H) Myotube survival (%) was determined by a MTT assay in myotubes treated with the conditions similar to ‘G’. (I) SIRT-1 mRNA (left side) and protein (right side) levels were determined in total mRNA and cell lysates, respectively. (J) PARP-1 acetylation levels were estimated from immunoblots after IP with the conditions similar to ‘G’. (K) PGC-1α acetylation levels were determined in total cell lysates from either myotubes treated with H2O2 or non-treated myotubes with or without PJ34 or RSV by immunoblotting after IP. Global cellular protein PARylation (L) or myotube survival (%) was determined in total cell lysates from either myotubes treated with H2O2 or non-treated myotubes with or without resveratrol. IP, immunoprecipitation; RSV, resveratrol.
Activated-PARP-1 inhibits SIRT-1 activity by depleting intracellular NAD+ levels and poly (ADP-ribosyl)ation
We next studied the molecular mechanism by which over activation of PARP-1 inhibits SIRT-1 function. Since SIRT-1 and PARP-1 share the same nuclear NAD+ pool, it is possible that one enzyme may influence the other's activity through competition for NAD+ [35]. Therefore, we measured the intracellular NAD+ content in myotubes treated with H2O2 and found that the treatment significantly decreased intracellular NAD+ content; however, inhibition of PARP-1 by either PJ34 or siRNAs blocked H2O2–induced NAD+ depletion (Fig. 5A). Although the depletion of intracellular NAD+ levels by PARP-1 inhibits SIRT-1 activity, it is not known whether PARP-1 also inhibits SIRT-1 activity in skeletal muscles by PARylation. Thus, we conducted IP assays using nuclear extracts from H2O2-treated myotubes with an antibody specific for PARylated proteins followed by immunoblot assays with an anti-SIRT-1 antibody. As shown in F igure 5B, H2O2 treatment had a marked increase in the level of SIRT-1 PARylation (Fig. 5B). Likewise, IP assays with an antibody specific for SIRT-1 followed by immunoblot assays with an anti-PAR antibody confirmed the H2O2-induced PARylation of SIRT-1 (Fig. 5C). However, reatment of myotubes with PJ34 or PARP-1-targeted siRNAs reversed the effect of PARP-1 on SIRT-1 PARylation (Fig. 5B and C), indicating that over-activation of PARP-1 inhibits the activity of SIRT-1 by both depleting intracellular NAD+ and PARylating SIRT-1. Next, we determined whether over activation of SIRT-1 could rescue myotubes from H2O2/PARP-1-induced cell death. Towards this end, we treated myotubes with resveratrol, which significantly increased SIRT-1 activity (Fig. 5D). Activation of SIRT-1 by resveratrol reduced PARP-1-mediated SIRT-1 PARylation (Fig. 5E). To explore the role of SIRT-1 in myotube survival, we isolated primary myoblasts (i.e. satellite cells) from SIRT-1 conditional (floxed) knockout mice (SIRT-1loxp/loxp) and infected these cells with AAV transducing either GFP or Cre (Fig. 5F). Infection of the floxed primary myoblasts with Cre, but not with GPF significantly reduced the activity of SIRT-1 (Fig. 5G). Activation of PARP-1 in AAV-Cre-infected myotubes by H2O2 significantly increased global protein PARylation (Fig. 5H) and decreased the survival of myotubes (Fig. 5I); however, treatment of these myotubes with resveratrol did not reverse the effect of H2O2 (Fig. 5H and I). These results indicate that H2O2-induced PARP-1 activation induces myotube death via inhibition of SIRT1 activity due to depletion of intracellular NAD+ and increased SIRT-1 PARylation.
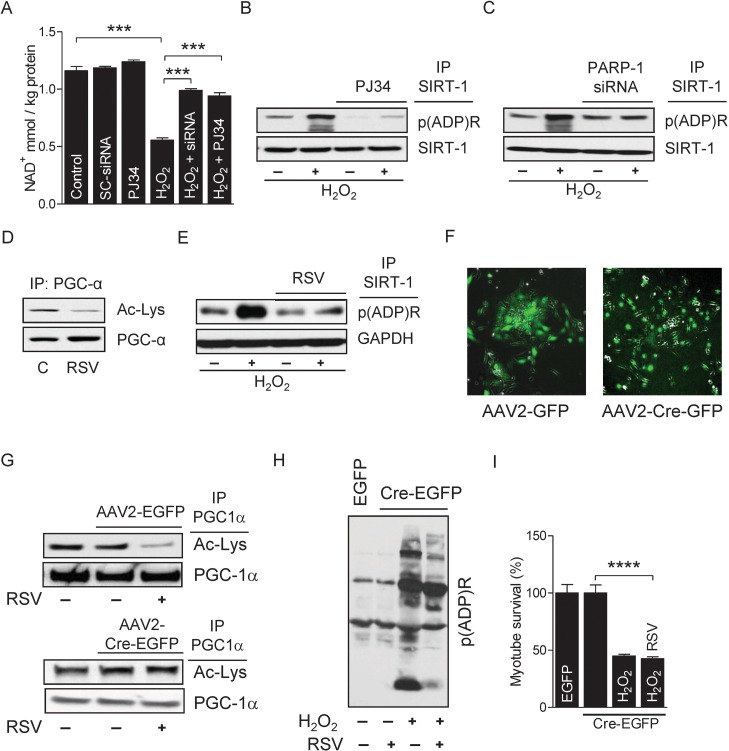
Figure 5. PARP-1 inhibits SIRT-1 activity by PARylation (A) NAD+ levels were determined in myotubes either in the presence or in the absence of H2O2 with or without the inhibition of PARP-1 by PJ34 or siRNA. (B) NAD+ levels were determined in myotubes either in the presence or in the absence of H2O2 with or without the inhibition of PARP-1 by PJ34 or siRNA. (B and C) SIRT-1 protein PARylation was determined in total cell lysates from myotubes treated with the conditions similar to ‘A’ by immunoblotting after IP. (D) PGC-1α acetylation levels were determined by immunoblotting in total cell lysates from myotubes treated with or without resveratrol. (E) SIRT-1 protein PARylation was determined in myotubes either in the presence or in the absence of H2O2 with or without RSV. (F) Infection of myotubes isolated from conditional knockout mice (flox/flox) with AAV2-GFP or AAV2-Cre-GFP to determine SIRT-1 knock-down. (G) PGC-1α acetylation levels were determined in total cell lysate from myotubes infected with either AAV2-GFP or AAV2-Cre-GFP in the presence or absence of RSV by immunoblotting after IP. Global cellular protein PARylation (H) and percentage of myotube survival (I) were determined in either AAV2-GFP or AAV2-Cre-GFP infected myotubes in the presence or in the absence of H2O2 or RSV. AAV, adeno-associated virus; GFP, green fluorescence protein; IP, immunoprecipitation; RSV, resveratrol.
Discussion
Skeletal muscle undergoes a profound age-related deterioration called sarcopenia, which is characterized by a marked decline in muscle mass and function. Although the pathogenesis of sarcopenia is complex, ROS accumulation stemming from mitochondrial dysfunction plays a key role in this process. PARP-1 is a central mediator of the response to cellular stress caused by physiological stressors, such as ROS and inflammation. While the basal activity of PARP-1 is necessary to maintain genome integrity and cellular homeostasis in response to oxidative stress, over-activation of PARP-1 induces a skeletal muscle decline that is more common in aged individuals than in younger people. Therefore, it is possible that regulation of skeletal muscle protein PARylation by PARP-1 would be different in aged people than that in younger individuals. Here, we demonstrate that in aged mice, exercise over-activates PARP-1 via GCN5-dependent acetylation in skeletal muscle and that activated-PARP-1 depletes cellular NAD+ levels, thereby inhibiting SIRT-1 activity that resulted in reduced mitochondrial biogenesis and metabolism, and increased the onset of fatigue and reduced the ability to produce and work. In contrast, SIRT1 has the ability to bind with and deacetylate PARP-1, and as a result of reduced PARP-1 activity, there is an increase in mitochondrial content and biogenesis in skeletal muscle from young mice. Interestingly, inhibition of PARP-1 in aged mice reduced the exercise-induced over activation of PARP-1 and promoted mitochondrial biogenesis via the SIRT-1/PGC-1α pathway, which resulted in an increased onset of fatigue an a decreased ability to produce total muscle work, suggesting that activation of SIRT-1 and/or inhibition of PARP-1 would be an effective way to improve muscle function in aging. As PJ34 delayed the onset (although not the extent) of fatigue, and it also improved the capacity to sustain the higher forces over the duration of the 360 contractions in our experiments, the ability to produce and sustain ATP production and delivery from the mitochondria to the muscles was presumably better when PARP-1 was inhibited. This speculation is consistent with observations of greater signaling for mitochondrial biogenesis (which would be the primary source of ATP generation) when PARP-1 was inhibited. It would have been interesting to test if the PJ34 would have markedly lowered the muscle fatigue rate as compared to control muscles throughout a sustained effort, if the muscles in both treatment groups had been required to maintain an identical absolute submaximal load over the course of the experiment (although this was not tested in the current study).
SIRT-1 has emerged as a major therapeutic target for aging and age-associated diseases, including sarcopenia [44, 45]. Therefore, understanding how oxidative stress modulates SIRT-1 is crucial to unraveling the mechanisms underlying sarcopenia. SIRT-1 has also been shown to protect skeletal muscle against ROS-induced muscle damage [46]. One interesting approach to ameliorating age-related skeletal muscle disorders would be to elevate intracellular NAD+ content, thereby activating the NAD+-dependent enzyme SIRT-1. In line with this, several studies have shown that increasing NAD+ levels in old mice restored mitochondrial function and decreased lactate production, reversing a pseudo-hypoxic state [47], which may be mTOR-dependent [48, 49]. In conjunction with increasing cellular NAD+ content, hindering other NAD+-dependent proteins would also be expected to enhance SIRT-1 activity. For example, in the present study, we showed in vitro and in vivo that inhibition of PARP-1, a major cellular NAD+ consumer, increased cellular NAD+ levels, which increased SIRT-1 activity, thereby augmenting mitochondrial content and biogenesis. We also showed that over-activation of PARP-1 by H2O2 increased global protein PARylation and depleted intracellular NAD+ content leading to myotube death. This suggests that one enzyme may influence the other's activity through competition for NAD+ and that a tight regulation of PARP-1 activity is important for cell survival. Our present work supports this concept by showing how the attenuation of PARP-1 increased intracellular NAD+ levels and enhanced SIRT-1 activity. This effect prompted the deacetylation and activation of the key metabolic transcriptional regulator PGC-1α, leading to increased mitochondrial content and biogenesis. Our data provide strong support for the idea that in aging skeletal muscles, over-activation of PARP-1 limits NAD+ availability for SIRT-1 function. This concept derives from the variation in the KM and kcat/KM of both enzymes for NAD+, which indicates that PARP-1 is more rapid and a more efficient NAD+ consumer than is SIRT1 [50]. Hence, it is possible that PARP-1 activity modulates NAD+ and this regulates SIRT1 function. While earlier data have reported that exercise increases SIRT1-activity [51]and other studies had speculated on a link between PARP-1 and SIRT-1 activities [32, 35], our study expands the consequences of this link to sarcopenia. In the present study, we demonstrated that the exercise-induced activation of PARP-1 significantly reduced SIRT-1 activity due to depletion of cellular NAD+ content and as a result increased skeletal muscle fatigue in aged mice. Interestingly, in vivo inhibition of PARP-1 by PJ34 blocked PARP-1 activity resulting in higher SIRT-1/PGC-1α activity and mitochondrial content and biogenesis in skeletal muscle from aged mice. More importantly, inhibition of PARP-1 significantly improved muscle performance in aged mice. Moreover, activation of SIRT-1 by resveratrol treatment mimicked the effects of in vivo PARP-1 inhibition in myotubes. In agreement with the findings of our present study, our earlier results have shown that resveratrol appears to have modest therapeutic benefits for improving muscle mass in aged animals [38, 52, 53]. These results suggest that the activation of SIRT-1 by either modulating NAD+ availability or inhibiting other NAD+ consumers, such as PARP-1, could be an alternative means to activate SIRT-1 and perhaps to ameliorate, at least in part, the pathogenicity of sarcopenia.
Skeletal muscle abundantly expresses PARP-1, especially in response to oxidative stress [32]. In the present study, we demonstrated the mechanism of PARP-1 regulation in skeletal muscle. More precisely, treatment of myotubes with H2O2 or exercise in skeletal muscle, especially in aged mice, robustly activated PARP-1, as evidenced by enhanced PARylation of skeletal muscle proteins in addition to an enhanced acetylation of PARP-1 protein, indicating that acetylation of PARP-1 had indeed contributed to the increased PARP-1 activity. This is consistent with previous studies, which reported that treatment of cardiomyocytes with H2O2 increased both the acetylation level and the activity of PARP-1 [36]. Furthermore, data from our present study suggest that skeletal muscles from young and aged mice differentially regulate protein PARylation and that PARP-1 might be one of the downstream targets of the stress stimuli that initiate sarcopenia. Although many members of class I HDACs deacetylate PARP-1, in the present study, we found that SIRT-1 was also capable of deacetylating PARP-1, which is consistent with the results of a previous study in cardiomyocytes [37]. Our in vitro study further confirm that a deletion of the full catalytic core domain of SIRT-1, which eliminated its deacetylase activity, did not affect its binding to PARP-1; however, the deletion failed to suppress skeletal muscle protein PARylation. Introduction of resveratrol had no effect on the level of protein PARylation, indicating that the catalytic activity, and not the protein binding ability of SIRT-1, was necessary for blocking PARP-1 activity. These results also corroborate that deacetylation of PARP-1 in skeletal muscles is unique to SIRT-1, because the deletion of exon 4 of the SIRT-1 gene is unique to the SIRT-1 catalytic core domain rather than to other members of sirtuin family. This observation provides evidence for SIRT-1-dependent inactivation of PARP-1 in muscle. In line with our current findings, previous studies have shown that the acetyltransferase p300/CBP acetylates PARP-1 and increases its ability to regulate NF-κB-dependent gene transcription [39]. Another study has shown that the acetyltransferase PCAF acetylates PARP-1 [37]. Although, these two studies identified different lysine residues with different rates of acetylation by p300/CBP and PCAF, one of the PARP-1 fragments (aa 477 to 525 segment) was highly acetylated by both p300/CBP and PCAF. This suggests that PCAF and p300/CBP may have different preferences for lysine residues within PARP-1. In the present study, we identified GCN5 as a new acetyltransferase and activator of PARP-1. With a series of IP assays, we showed that in young mice, electrically evoked isometric exercise significantly decreased the acetylation level of PARP-1 due to an increased association with SIRT-1 and dissociation from GCN5. In contrast, the exercise-induced acetylation of PARP-1 was greatly increased in aged mice due to decreased association with SIRT-1 and increased association with GCN5. These results indicate that dysregulation of SIRT-1 may be a primary mechanism that regulates the over-activation of PARP-1 in the skeletal muscles of aged mice. Furthermore, it was logical to anticipate that GCN5 could act as an acetyltransferase of PARP-1, because GCN5 has been shown to acetylate other skeletal muscle proteins in response to a variety of stimuli [54] and it is 70% identical to PCAF [55], which is a p300/CREB binding protein/associated factor. Therefore, it is reasonable to expect that GCN5, PCAF, and p300/CBP may control PARP-1 activity differently in different cellular and physiological conditions. It will be interesting to determine which lysine residues within PARP-1 are acetylated by GCN5 in skeletal muscle.
Many lines of evidence have shown opposing roles of PARP-1 and SIRT-1 for the same target. For example, while PARP-1 increases p53 nuclear translocation and its transcriptional activity via PARylation, SIRT-1 deacetylates p53 and inhibits its transcriptional activity [56, 57]. Similarly, PARP-1 and SIRT-1 regulate other targets such as NF-κB, FOXO, and Ku70 [57-59] in opposite directions. Thus, in order for the activity of one of the two enzymes to pre-dominate, it is necessary to inhibit the activity of the other. The data in the present study demonstrate that both PARP-1 and SIRT-1 have the capability to counterbalance each other's activity in young and aged mice. The model proposed in Fig. 6 depicts how SIRT-1 and PARP-1 may regulate each other's activity in skeletal muscle between young and aged mice to affect mitochondria abundance thereby modulating muscle fatigue and potentially impacting sarcopenia.
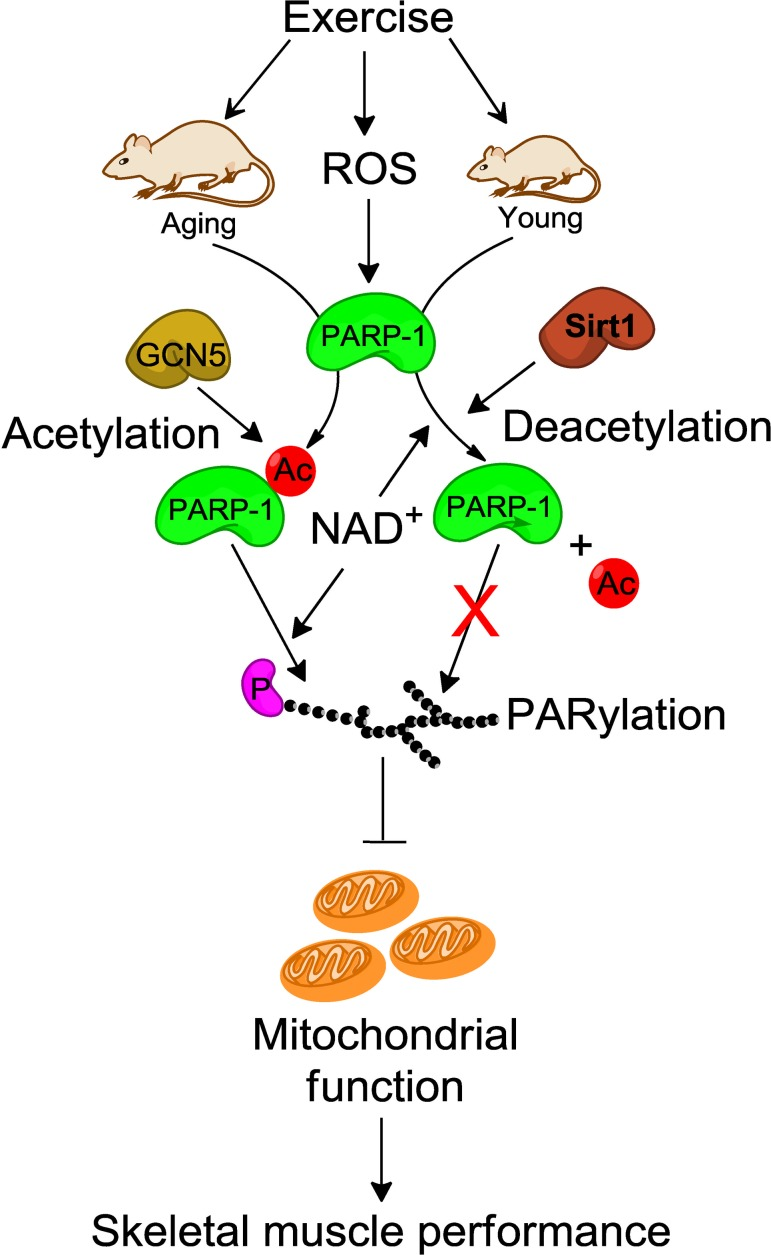
Figure 6. Schematic illustration of how SIRT-1 may protect skeletal muscle from PARP-1-induced muscle fatigue In young mice, SIRT-1 inhibits the exercise-induced PARP-1 activity by deacetylation-dependent mechanism and as a result increases skeletal muscle performance via enhanced mitochondrial biogenesis. In contrast, dysregulation of SIRT-1 in skeletal muscle from aged mice reduces skeletal muscle performance due to higher PARP-1 activity via GCN5 mediated acetylation. ROS, reactive oxygen species; NAD, nicotinamide adenine dinucleotide; Ac, acetylation; P, protein.
In summary, we demonstrated that ROS-induced PARP-1 activation decreased myotube survival via depleting cellular NAD+ levels, and that depletion inhibits SIRT-1 activity. We also demonstrated a new mode of PARP-1 activation by GCN5-mediated acetylation in skeletal muscle. SIRT-1 has the ability to physically bind to PARP-1 and deacetylate it, and that effect results in suppression of PARP-1 enzymatic activity in skeletal muscle from young mice following exercise. This protective role of SIRT-1 was suppressed in skeletal muscle of aged mice, but this suppression was reversed by PARP-1 inhibition. These data provide strong evidence for the existence of a functional interplay between PARP-1 and SIRT-1 and give novel insights into the modulation of skeletal muscle fatigue by SIRT-1 in sarcopenia.
Methods
Animals
The Institutional Animal Care and Use Committee from the West Virginia University School of Medicine approved all experimental procedures. A total of 34 mice were used in these experiments. These include 3 month old young (n=16), and 26 months old aged (n=18) male C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME). All mice were kept in a temperature-controlled room on a 12-h light/dark cycle, with 60% humidity, and food and water ad libitum. Mice were anesthetized with 2% isoflurane gas using a small animal anesthetic system (Surgivet Anesco Inc., Waukesha, WI.) before electrically-evoked contractions (exercise) or terminal surgery.
Electrically-evoked isometric contractions
In situ electrically stimulated isometric contractions were conducted on a custom-built mouse dynamometer as described previously [9]. Briefly, mice were anesthetized with a mixture of oxygen (97%) and isoflurane gas (3%) and placed on a plate that was heated to 37°C. The right ankle was positioned at 90°C of flexion and was aligned with the axis of rotation of the servomotor (Model 6350* 350; Cambridge Technology, Scientific, Aurora, ON, Canada). The foot was secured to the footplate that was connected to the servomotor. Commercially available software (Dynamic Muscle Control; Aurora Scientific, Aurora, ON, Canada) was used to control the servomotor providing for the angular position of the foot.
Muscle contractions of the plantar flexor muscles were stimulated via subcutaneous platinum electrodes that were placed on either side of the tibial nerve near the popliteal fossa. Electrode placement was tested via a short stimulation of the nerve to cause plantar flexion twitches. When stimulated, the foot plantar flexed without any visible appearance of eversion, or inversion, of the foot. Twenty electrically evoked (10-V, 100-Hz, 200 μs pulses) isometric contractions of the plantar flexor muscle group were obtained in one limb. Each contraction train lasted for 3 s, and a 10-s recovery period occurred between subsequent contractions. The contralateral limb served as the intra animal control. Muscle functional data were collected as a force×time curve during isometric contractions for each session and values were normalized to each animal's body weight. Muscle fatigue data were assessed over 360 contractions at 40Hz (0.3s duration with 200 μs pulses). The fatigue index (expressed as percent of the starting force) was calculated as: [the first contraction – desired contraction)/first contraction x 100]. The contractile data were analyzed offline (Dynamic Muscle Analysis software; Aurora Scientific).
Cell Culture
Isolation of primary myoblasts from SIRT-1flox/flox mice was performed as described previously [60]. Before starting the experiments, myoblasts from passages 5 and 6 were cultured in growth medium (GM; DMEM containing 20% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin), and differentiation was induced by replacing GM with differentiation medium (DM; GM containing 2% horse serum instead 20% fetal calf serum) when they were 70-80% confluent. Myotubes maintained in DM for four days were used in all experiments.
siRNA transfection
For siRNA-mediated knockdown studies, myotubes were transfected with 500 pmol of siRNA specific for mouse PARP-1 or nonspecific siRNA (Santa Cruz Biotechnology). RNA transfection studies were performed with Lipofectamine RNAi MAX (Invitrogen) according to the manufacturer's instructions. After 8 h, the transfection medium was replaced with DM. Subsequent assays were conducted 24 to 48 h after transfection.
Cell survival assay
Fresh DM was added to both treated and non-treated myotubes cultured in 24-well plates followed by incubation with 100 μl MTT solution (10 mg/ml in PBS) for 4 h in a cell culture incubator. One hundred microliters of lysis buffer (20% SDS in 50% dimethyl formamide, pH 4-7) was added to each well, and then the plates were further incubated for 16 h in a cell culture incubator. The absorbance was read at 570 nm using a microplate reader (DynexTechnologies Limited, Worthing, West Sussex, UK). Wells containing DM without myotubes were included as a blank.
Cellular NAD+
NAD+ was estimated colorimetrically using an NAD/NADH assay kit (Abcam, Cambridge, MA) as described previously [61]. The standard was prepared according to the manufacturer's protocol. The NAD/NADH ratio was calculated using the formula: NADt (NAD and NADH) — NADH/NADH and was expressed as mM/kg protein.
Reverse transcription and quantitative PCR
Real-time RT-PCR was performed as described previously (Mohamed et al. 2013). The relative amounts of amplified transcripts (2−△CT) were estimated by the comparative CT (−△CT) method and normalized to an endogenous reference (GAPDH) relative to a calibrator. All PCR products were verified on an agarose gel stained with ethidium bromide to discriminate between the correct amplification products and potential primer dimers. The primers used in this study are described previously [61].
Immunoblots
Total cell lysate extraction from either myotubes or skeletal muscles and westernblot were performed as described previously [60]. Anti-p(ADP)R (sc-56198) and anti-PGC-1α(sc-13067) were purchased from Santa Cruz Biotechnology. Anti-PARP-1 (9542), anti-GAPDH (5174), anti-acetylated lysine (9441) and anti-GCN5 (3305) were purchased from Cell Signaling.
In vitro pull-down assay
Protein-protein interactions and protein acetylation levels were determined by immunoprecipitation (IP) using 150 μg protein and A/G-agarose beads as described previously [60, 61].
Statistical analysis. The results are expressed as the means ± SEM. Comparisons among different groups were performed by one-way ANOVA followed by Bonferroni post-testing. Paired data were evaluated by Student t test. A P value of < 0.05 was considered statistically significant. Each experiment was repeated at least three times using three different mice.
Acknowledgments
We would like to acknowledge the West Virginia University Microscope Imaging Facility, which is supported by the Mary Babb Randolph Cancer Center and NIH grant 5P20RR016440, P30RR032138/-GM103488 and P20RR016477, and NIH Grant P20GM103434 to the West Virginia IDeA Network for Biomedical Research Excellence.
Conflicts of Interest
No conflict of interest exits for any author.
References
- 1. Welch AA, MacGregor AJ, Minihane AM, Skinner J, Valdes AA, Spector TD, Cassidy A. Dietary fat and fatty acid profile are associated with indices of skeletal muscle mass in women aged 18-79 years. J Nutr. 2014; 144: 327 -334. [PubMed] .
- 2. Prado CM, Wells JC, Smith SR, Stephan BC, Siervo M. Sarcopenic obesity: A Critical appraisal of the current evidence. Clin Nutr. 2012; 31: 583 -601. [PubMed] .
- 3. Rosenberg IH. Sarcopenia: origins and clinical relevance. J Nutr. 1997; 127: 990S -991S. [PubMed] .
- 4. Fielding RA, Vellas B, Evans WJ, Bhasin S, Morley JE, Newman AB, Abellan van Kan G, Andrieu S, Bauer J, Breuille D, Cederholm T, Chandler J, De Meynard C, et al. Sarcopenia: an undiagnosed condition in older adults. Current consensus definition: prevalence, etiology, and consequences. International working group on sarcopenia. J Am Med Dir Assoc. 2011; 12: 249 -256. .
- 5. Ghosh S, Lertwattanarak R, Garduno JD, Galeana JJ, Li J, Zamarripa F, Lancaster JL, Mohan S, Hussey S, Musi N. Elevated Muscle TLR4 Expression and Metabolic Endotoxemia in Human Aging. J Gerontol A Biol Sci Med Sci. 2014; .
- 6. Jang YC, Sinha M, Cerletti M, Dall'Osso C, Wagers AJ. Skeletal muscle stem cells: effects of aging and metabolism on muscle regenerative function. Cold Spring Harb Symp Quant Biol. 2011; 76: 101 -111. [PubMed] .
- 7. Garcia-Prat L, Sousa-Victor P, Munoz-Canoves P. Functional dysregulation of stem cells during aging: a focus on skeletal muscle stem cells. FEBS J. 2013; 280: 4051 -4062. [PubMed] .
- 8. Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, Perdiguero E, Munoz-Canoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014; 506: 316 -321. [PubMed] .
- 9. Ryan MJ, Jackson JR, Hao Y, Leonard SS, Alway SE. Inhibition of xanthine oxidase reduces oxidative stress and improves skeletal muscle function in response to electrically stimulated isometric contractions in aged mice. Free Radic Biol Med. 2011; 51: 38 -52. [PubMed] .
- 10. Fulle S, Protasi F, Di Tano G, Pietrangelo T, Beltramin A, Boncompagni S, Vecchiet L, Fano G. The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exp Gerontol. 2004; 39: 17 -24. [PubMed] .
- 11. Ji LL. Antioxidant signaling in skeletal muscle: a brief review. Exp Gerontol. 2007; 42: 582 -593. [PubMed] .
- 12. Vays VB, Eldarov CM, Vangely IM, Kolosova NG, Bakeeva LE, Skulachev VP. Antioxidant SkQ1 delays sarcopenia-associated damage of mitochondrial ultrastructure. Aging (Albany NY). 2014; 6: 140 -148. [PubMed] .
- 13. Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML. Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol Cell. 2013; 52: 272 -285. [PubMed] .
- 14. Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006; 7: 517 -528. [PubMed] .
- 15. Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010; 35: 208 -219. [PubMed] .
- 16. Woodhouse BC and Dianov GL. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair (Amst). 2008; 7: 1077 -1086. [PubMed] .
- 17. Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signal. Genes Dev. 2005; 19: 1951 -1967. [PubMed] .
- 18. Kraus WL. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol. 2008; 20: 294 -302. [PubMed] .
- 19. Ha HC and Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. 1999; 96: 13978 -13982. [PubMed] .
- 20. Ingram DK, Zhu M, Mamczarz J, Zou S, Lane MA, Roth GS, deCabo R. Calorie restriction mimetics: an emerging research field. Aging Cell. 2006; 5: 97 -108. [PubMed] .
- 21. Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009; 325: 201 -204. [PubMed] .
- 22. Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005; 310: 1641 [PubMed] .
- 23. Li Y, Xu W, McBurney MW, Longo VD. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008; 8: 38 -48. [PubMed] .
- 24. Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, Wright SM, Mills KD, Bonni A, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008; 135: 907 -918. [PubMed] .
- 25. Spindler SR. Caloric restriction: from soup to nuts. Ageing Res Rev. 2010; 9: 324 -353. [PubMed] .
- 26. Yu J and Auwerx J. Protein deacetylation by SIRT1: an emerging key post-translational modification in metabolic regulation. Pharmacol Res. 2010; 62: 35 -41. [PubMed] .
- 27. Vinciguerra M, Fulco M, Ladurner A, Sartorelli V, Rosenthal N. SirT1 in muscle physiology and disease: lessons from mouse models. Dis Model Mech. 2010; 3: 298 -303. [PubMed] .
- 28. Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003; 12: 51 -62. [PubMed] .
- 29. Rathbone CR, Booth FW, Lees SJ. Sirt1 increases skeletal muscle precursor cell proliferation. Eur J Cell Biol. 2009; 88: 35 -44. [PubMed] .
- 30. Machida S and Booth FW. Increased nuclear proteins in muscle satellite cells in aged animals as compared to young growing animals. Exp Gerontol. 2004; 39: 1521 -1525. [PubMed] .
- 31. Gurd BJ, Yoshida Y, McFarlan JT, Holloway GP, Moyes CD, Heigenhauser GJ, Spriet L, Bonen A. Nuclear SIRT1 activity, but not protein content, regulates mitochondrial biogenesis in rat and human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2011; 301: R67 -75. [PubMed] .
- 32. Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011; 13: 461 -468. [PubMed] .
- 33. Bai P, Canto C, Brunyanszki A, Huber A, Szanto M, Cen Y, Yamamoto H, Houten SM, Kiss B, Oudart H, Gergely P, Menissier-de Murcia J, Schreiber V, et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011; 13: 450 -460. [PubMed] .
- 34. Sauve AA, Moir RD, Schramm VL, Willis IM. Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol Cell. 2005; 17: 595 -601. [PubMed] .
- 35. Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005; 280: 43121 -43130. [PubMed] .
- 36. Adamietz P. Poly(ADP-ribose) synthase is the major endogenous nonhistone acceptor for poly(ADP-ribose) in alkylated rat hepatoma cells. Eur J Biochem. 1987; 169: 365 -372. [PubMed] .
- 37. Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009; 29: 4116 -4129. [PubMed] .
- 38. Ryan MJ, Jackson JR, Hao Y, Williamson CL, Dabkowski ER, Hollander JM, Alway SE. Suppression of oxidative stress by resveratrol after isometric contractions in gastrocnemius muscles of aged mice. J Gerontol A Biol Sci Med Sci. 2010; 65: 815 -831. [PubMed] .
- 39. Hassa PO, Haenni SS, Buerki C, Meier NI, Lane WS, Owen H, Gersbach M, Imhof R, Hottiger MO. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem. 2005; 280: 40450 -40464. [PubMed] .
- 40. Bejma J and Ji LL. Aging and acute exercise enhance free radical generation in rat skeletal muscle. J Appl Physiol (1985). 1999; 87: 465 -470. [PubMed] .
- 41. Gibson GE, Zhang H, Xu H, Park LC, Jeitner TM. Oxidative stress increases internal calcium stores and reduces a key mitochondrial enzyme. Biochim Biophys Acta. 2002; 1586: 177 -189. [PubMed] .
- 42. Schraufstatter IU, Hyslop PA, Hinshaw DB, Spragg RG, Sklar LA, Cochrane CG. Hydrogen peroxide-induced injury of cells and its prevention by inhibitors of poly(ADP-ribose) polymerase. Proc Natl Acad Sci U S A. 1986; 83: 4908 -4912. [PubMed] .
- 43. Virag L and Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002; 54: 375 -429. [PubMed] .
- 44. Tonkin J, Villarroya F, Puri PL, Vinciguerra M. SIRT1 signaling as potential modulator of skeletal muscle diseases. Curr Opin Pharmacol. 2012; 12: 372 -376. [PubMed] .
- 45. Joseph AM, Malamo AG, Silvestre J, Wawrzyniak N, Carey-Love S, Nguyen LM, Dutta D, Xu J, Leeuwenburgh C, Adhihetty PJ. Short-term caloric restriction, resveratrol, or combined treatment regimens initiated in late-life alter mitochondrial protein expression profiles in a fiber-type specific manner in aged animals. Exp Gerontol. 2013; 48: 858 -868. [PubMed] .
- 46. Pardo PS, Mohamed JS, Lopez MA, Boriek AM. Induction of Sirt1 by mechanical stretch of skeletal muscle through the early response factor EGR1 triggers an antioxidative response. J Biol Chem. 2011; 286: 2559 -2566. [PubMed] .
- 47. Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013; 155: 1624 -1638. [PubMed] .
- 48. Williamson DL. Normalizing a hyperactive mTOR initiates muscle growth during obesity. Aging (Albany NY). 2011; 3: 83 -84. [PubMed] .
- 49. Leontieva OV and Blagosklonny MV. M(o)TOR of pseudo-hypoxic state in aging: rapamycin to the rescue. Cell Cycle. 2014; 13: 509 -515. [PubMed] .
- 50. Smith BC, Hallows WC, Denu JM. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal Biochem. 2009; 394: 101 -109. [PubMed] .
- 51. Gurd BJ, Perry CG, Heigenhauser GJ, Spriet LL, Bonen A. High-intensity interval training increases SIRT1 activity in human skeletal muscle. Appl Physiol Nutr Metab. 2010; 35: 350 -357. [PubMed] .
- 52. Bennett BT, Mohamed JS, Alway SE. Effects of resveratrol on the recovery of muscle mass following disuse in the plantaris muscle of aged rats. PLoS One. 2013; 8: e83518 [PubMed] .
- 53. Jackson JR, Ryan MJ, Hao Y, Alway SE. Mediation of endogenous antioxidant enzymes and apoptotic signaling by resveratrol following muscle disuse in the gastrocnemius muscles of young and old rats. Am J Physiol Regul Integr Comp Physiol. 2010; 299: R1572 -1581. [PubMed] .
- 54. Gurd BJ. Deacetylation of PGC-1alpha by SIRT1: importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl Physiol Nutr Metab. 2011; 36: 589 -597. [PubMed] .
- 55. Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996; 382: 319 -324. [PubMed] .
- 56. Kanai M, Hanashiro K, Kim SH, Hanai S, Boulares AH, Miwa M, Fukasawa K. Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat Cell Biol. 2007; 9: 1175 -1183. [PubMed] .
- 57. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001; 107: 137 -148. [PubMed] .
- 58. Wong K, Zhang J, Awasthi S, Sharma A, Rogers L, Matlock EF, Van Lint C, Karpova T, McNally J, Harrod R. Nerve growth factor receptor signaling induces histone acetyltransferase domain-dependent nuclear translocation of p300/CREB-binding protein-associated factor and hGCN5 acetyltransferases. J Biol Chem. 2004; 279: 55667 -55674. [PubMed] .
- 59. Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004; 23: 2369 -2380. [PubMed] .
- 60. Mohamed JS, Lopez MA, Cox GA, Boriek AM. Ankyrin repeat domain protein 2 and inhibitor of DNA binding 3 cooperatively inhibit myoblast differentiation by physical interaction. J Biol Chem. 2013; 288: 34 24560 -24568. [PubMed] .
- 61. Mohamed JS, Hajira A, Pardo PS, Boriek AM. MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1alpha network in skeletal muscle. Diabetes. 2014; 63: 1546 -1559. [PubMed] .