



Introduction
Although adult hepatocytes are characterized by a very low replicative rate, they can rapidly re-entry into the cell cycle following tissue loss/death [1–4]. The best characterized experimental model to study liver regeneration consists of removal of 2/3 of hepatic parenchyma (2/3 PH) in rodents [5]. In response to 2/3 PH, the remnant liver cells proliferate until the tissue mass is recovered (within 7 to 10 days). The precise molecular mechanisms associated to G0/ to G1 transition are not completely understood. However, the earliest documented changes after 2/3 PH involve increased activity of urokinase plasminogen activator (uPA) and migration of Notch1 and β- catenin to hepatocyte nuclei within 15-20 minutes [2,6]. Then, uPA-mediated extracellular matrix remodeling determines release in blood circulation of active hepatocyte growth factor (HGF), embedded in ECM in an inactive form [7–9]. Within 30-60 minutes post 2/3 PH, activation of HGF and epidermal growth factor (EGF) receptors occurs [10]. Combined EGFR and MET signaling has been found fundamental to normal hepatocyte function and liver regeneration [11]. Accordingly, their simultaneous elimination affects both the processes [11]. Furthermore, soon after 2/3 PH, a rapid increase in blood concentration of norepinephrine, tumor necrosis factor (TNF)-α, interleukin (IL) 6, serotonin, and bile acids take place [1–3,12–15]. Though no directly mitogenic, these factors orchestrate and optimize the timing and intensity of intracellular signals essential for controlling hepatocyte proliferation and paracrine cell interactions [6]. Prior to DNA synthesis, activation and nuclear translocation of transcription factors, such as signal transducer and activator of transcription 3 (STAT3), CCAAT/enhancer-binding protein beta (C/EBPβ) and nuclear factor kappa B (NFκB), occurs [2]. Enhanced expression of cell cycle inhibitors (p21 and p53), immediate early genes (IEGs) (c-Fos, c-Jun and c-Myc) transforming growth factor (TGF)-β [1] and of the transcription factors Octamer 4 and Nanog [16] are also observed. All these events lead to transcription of delayed early genes, coding for cell cycle regulatory proteins, namely cyclins [17–19]. microRNAs have also been involved in the regulation of hepatocyte DNA synthesis [20]. As an example, microRNA-21 has been found to accelerate cyclin D1 translation and cell-cycle progression during mouse liver regeneration [21]. Being liver regeneration a very strictly controlled process, hepatocyte replication ceases once the liver weight/body weight ratio has regained its original values [6,22].
Based on these findings, liver regeneration has been divided into three phases: i) priming, characterized by growth factors activation and cytokine release, ii) proliferation, promoted by immediate early gene/transcription factor activation, and iii) termination, likely governed by signal transduction pathways, such as the TGF-β-TGFβR-mediated pathway [2].
Since aging affects the regenerative response of the liver after chronic tissue injury or following surgical resection [23], it represents a critical problem in aged patients with liver disease. The first studies focusing on the effect of aging on liver regeneration date back to more than 50 years ago. At that time, Bucher et al. [24] found that the regenerative response, though preserved, was considerably reduced and retarded in aged rodents after 2/3 PH. Several subsequent studies confirmed that observation [25–29]. Despite several works focused on the subject, the molecular mechanisms underlying the age-dependent impairment of the liver regenerative capacity remain elusive.
Intra-cellular factors affecting liver regeneration in aged rodents
Several different explanations have been proposed to justify the decline of the liver regenerative potential with age. Epigenetic alterations (i.e. histone deacetylation, methylation of gene promoter sequences and chromatin remodeling) are considered to be critical as they lead to modified expression/activation/action of genes related to hepatocyte proliferation (i.e. Foxm1b, Cdc2, c-Myc, c-Fos, c/EBP) [27,30,31].
About 20 years ago, Wang et al. [30] showed that reduced expression of the gene coding for the Forkhead Box M1B (FoxM1B) contributed to the proliferation defects observed in aged liver. FoxM1B is an ubiquitously expressed transcription factor restricted to proliferating cells of the mouse embryo (including liver) whose expression diminishes during postnatal cellular differentiation [32]. Liver regeneration is associated to FoxMB1 reactivation prior to S phase and sustained throughout the period of hepatocyte proliferation [32]. A positive correlation between age-related cell proliferation defects and diminished expression of FoxM1B and of its cell cycle-associated target genes has been demonstrated [33–35]. In addition, Wang et al. [30] found that FoxM1B gene overexpression in livers of old transgenic mice induced a regenerative response that was similar to that of young mice. Restoration of the young regenerating liver phenotype was associated with increased expression of several cell cycle-related genes (i.e. Ccnd1, Ccna2, Ccnf, Ccnb1, Ccnb2, coding for cyclin D1, cyclin A2, cyclin F, cyclin B1, cyclin B2, respectively). Moreover, co-transfection assays showed that FoxM1B activated transcription of cyclin B1 and cyclin D1 promoters, suggesting that those genes were direct targets of the transcription factor [30]. Collectively, all these data demonstrated that FoxM1B controls the transcriptional network of liver cell proliferation-associated genes.
Few years later, Iakova et al. [27] identified the transcription factor C/EBPα as a major contributing factor in the reduced regenerative response of aged mice to PH. In their study, Iakova et al. [27] demonstrated that in aged livers C/EBPα, a strong inhibitor of cyclin-dependent kinases (cdks) highly expressed in rodent liver [36–39], formed a C/EBPα-Rb-E2F4 complex. This complex bound to and repressed the E2F-mediated transcription of the gene c-Myc, which plays a central role in liver regeneration [15,40]. On these bases, authors hypothesized that post PH in young livers cdk2 is detached from C/EBPα by cyclins E and A, which bound to and activated the kinase. This, in turn, phosphorylates Rb blocking the repression of the c-myc promoter. On the opposite, the switch of the C/EBPα activity from cdk inhibition to repression of E2F-mediated transcription prevents old livers from eliminating the C/EBP-mediated growth inhibition post-PH [27]. It was also suggested that the switch could be related to the age-dependent increase of Brm, a chromatin remodeling protein found to interact with C/EBPα in aged livers [41,27]. In this context, it should be noted that Brm and cdk2 interact with the same region of C/EBPα [38,41]. All together, these findings led Iakova and colleagues to hypothesize that Brm might replace cdk2 in old livers leading to initiation of the C/EBPα-Rb-E2F4 complex formation and to recruitment of C/EBPα in the E2F promoters [27].
An additional hypothesis was proposed by Gagliano et al. [31] who analyzed the cell cycle gene expression in the regenerating livers of young and old rats 24 hours after CCl4 administration. They found that in aging livers the regenerative response was associated to a reduction of c-Fos, c-Myc, Transforming growth factor-a (TGF-α) and Heat shock protein 70 (HSP70) mRNA levels, while Hgf mRNA levels were found to be increased [42,43]. However, several controversial data on the expression profiles of cell cycle related genes in regenerating livers of young and aged mice have been reported. Indeed, while Enkhbold et al. [44], found decreased Hgf, Met, Ccnd1 and Ccna2 mRNA levels in aged compared to young mouse livers, more recently Pibiri et al. [45] did not observe major age-dependent changes in the expression of IEGs (c-Jun, c-Fos and Egr-1), genes coding for cytokines/growth factors (Tnf-α,Il-6, Hgf, Tgf-α) or transcription factors (NF-κB, Stat3 and AP-1) [3,21,22]. Nevertheless, in the same study Ccnd1 gene, coding for the G1-phase protein cyclin D1, was found to be significantly down-regulated in the liver of aged mice.
Overall, the role of the early events involved in cell cycle regulation and impairment of liver regeneration in aged hepatocytes still remains elusive.
Recent studies focusing on the liver regenerative capacity of aged animals have underlined a fundamental role for intra-cellular factors involved in cell adhesion and in circadian genes regulation.
Role of factors involved in cell adhesion: BubR1 and YAP
BubR1 protein regulates the spindle assembly checkpoint and is involved in cellular senescence
Reduction in the hepatic levels of the mitotic checkpoint protein BubR1 (budding uninhibited by benzimidazole-related 1) has been suggested to be crucial in the age-dependent impairment of liver regeneration [46].
BubR1 plays an important role in the spindle assembly checkpoint to prevent chromosome unequal segregation during mitosis, thus preserving chromosomal stability [47]. BubR controls the anaphase-promoting complex, or cyclosome, (APC), a large E3 ubiquitin ligase, until all kinetochores are suitably attached to microtubules. When all the kinetochores establish bipolar attachment, APC degrades securin which, in turn, activates the separase. Then, proteolysis of the cohesion complex by separase triggers the onset of anaphase [47,48].
Several studies have also suggested a role for BubR1 in regulating aging. Indeed, decreased BubR1 expression causes cellular senescence through up-regulation of the cell cycle inhibitor p16INK4a. Moreover, mutant mice with decreased BubR1 expression (10% of the normal level) display various progeroid phenotypes [49–53].
Low-BubR1-expressing mutant (BubR1L/L) mice display delayed liver regeneration following PH
Ikawa-Yoshida and colleagues [46] analyzed liver regeneration after 2/3 PH in Low-BubR1-expressing mutant (BubR1L/L) mice characterized by a 20% BubR1 reduction. These mice do not display any severe phenotype during growth and development [53], therefore providing a useful model to investigate the role of BubR1 in response to various kinds of stress. Analysis of BubR1 gene expression in intact livers of C57Bl/6JJcl normal mice revealed a significant decrease of BubR1 mRNA levels in aged compared to young livers [46], confirming the inverse correlation between aging and BubR1 expression. Furthermore, in BubR1+/+ wild type animals mRNA levels of BubR1 significantly increased post-PH, suggesting the involvement of the protein in the regenerative response. Accordingly, BubR1L/L mice displayed a delayed liver regeneration post-PH which was associated to increased hepatocellular necrosis and intercalated disc anomalies. The observed structural intercellular alterations consisted of widened inter-hepatocyte and peri-sinusoidal spaces, smaller hepatocytes and early-stage microvilli atrophy. These changes were associated to the BubR1-dependent reduction of the expression of desmocollin-1 (DSC1), a desmosome transmembrane cell adhesion protein, highly expressed in the liver [54].
Based on these findings, the authors proposed the following key role for BubR1 in regulating microstructural adaptation during liver regeneration. To prevent an excessive detachment-induced cell death (DICD), due to portal hypertension consequent to vasculature reduction following PH [55], only a transient DICD associated to G1 arrest occurs. The increased BubR1-mediated DSC1 expression prevents massive DICD and anoikisis, and, thus, hepatocyte focal necrosis. Indeed, DSC1 causes desmosomes reinforcement to maintain proper cell attachments. On the opposite, in conditions associated to low BubR1 expression, reduction of DSC1 is responsible for the weakened microstructural adaptation post-surgery, thus leading to hepatocyte focal necrosis. Support to these findings stems from the work of Collins et al. [56] who demonstrated that impaired cell attachment caused DICD and G1 arrest in mammary epithelial cells in a p21-dependent manner. In keeping with these results, Ikawa-Yoshida and al [46]. suggested that the delayed liver regeneration observed in (BubR1L/L) mice was related to p21 up-regulation, rather than to a direct effect of low BubR1 expression.
Summarizing, findings here reported attributed the decline of the regenerative response in aged liver to BubR1 reduction which leads to decreased expression of DISC1, a cell adhesion protein involved in tissue reconstitution after damage. Yes-associated protein (YAP).
Yes-associated protein (YAP)
Interestingly, BubR1 expression has been found to be up-regulated by YAP (57), a downstream effector of a complex network of proteins named the mammalian tumor suppressor Hippo signaling pathway [58,59]. Hippo pathway controls organ size via regulation of cellular proliferation, survival and differentiation. These actions are mediated by Hippo-dependent inactivating phosphorylation and cytoplasmic retention of the transcriptional co-activators YAP (Yes-associated protein) and TAZ (transcriptional co-activator with PDZ binding motif). The most relevant residues that keep YAP and TAZ inhibited are represented by serine (S)127 and S381 in YAP and S89 and S311 in TAZ [60]. In case of loss of Hippo function, YAP and TAZ move to the cell nucleus and associate with various DNA-binding proteins, e.g. TEAD factors, driving gene transcription [61,62]. In rodent livers YAP is involved in proliferative signals, and its increased expression causes hepatomegaly and, when sustained, HCC development [62–65].
YAP protein controls transcription of spindle checkpoint genes through physical association with BubR1
Recent studies of Yang et al. [66,57] have shown that in mammalian epithelial cells the mitotic phosphorylation of YAP by CDK1, mainly in tyrosine (T)119 and S289 residues, ensured the spindle check point activation, responsible for correct chromosome segregation and mitosis. Furthermore, overexpression of mitotic phosphorylated YAP determined dysregulation of the spindle checkpoint, failure in maintaining normal mitosis and genomic integrity, and oncogenesis [66,57]. Mechanistically, the Authors have documented that the mitotic phosphorylated form of YAP controls transcription of spindle checkpoint genes through physical association with BubR1 [57]. Notably, mitotic phosphorylation of YAP was also required for BubR1 transcription [57]. Thus, BubR1 was suggested to be a transcriptional target of YAP in mediating spindle checkpoint function.
Given the role of BubR1 in regulating cell adhesion and liver regeneration [46], its YAP-dependent transcriptional activation appears very interesting. Indeed, similarly to BubR1, YAP is involved in cytoskeleton and cell adhesion regulation [67] and its reduced activation has been correlated to liver regeneration impairment during aging [68,45].
YAP protein is regulated by mechanical signals
Several studies have documented YAP involvement in mechanical signaling. Indeed, cell density [69], cell adhesion [69], cell morphology [70], ECM rigidity [71], and mechanical cell stress [72] have been identified as regulators of YAP/TAZ activity [67,73,74]. Support to this notion stems from the following findings: i) in low cell density culture YAP is predominantly localized to the nucleus in its active un-phosphorylated form while in high density culture is phosphorylated and localized in the cytoplasm [69]; ii) in cells grown on small domains YAP is mostly cytoplasmic, whereas it is localized in the nucleus on large domains [70]; iii) on hard substrates, YAP and TAZ are predominantly nuclear while increasingly accumulate in the cytoplasm on softer substrates [71]; iv) mechanical stretching of contact inhibited cells induces YAP/TAZ re-entry into the nucleus to stimulate cellular proliferation [72].
Collectively, all these studies have identified actin cytoskeletal reorganization as a dominant regulator of YAP and TAZ activity [73]. Studies suggesting a functional connection between G protein-coupled receptor (GPCR)/Rho signaling, cytoskeletal reorganization and YAP/TAZ activity support this finding [75–77]. In particular, Yu et al. [75], have reported that activation of GPCRs by chemical stimuli regulates YAP activity depending on the particular G protein coupled to the receptor. Indeed, G-protein binding may cause increase or inhibition of YAP function by enhancing or inhibiting the activity of the YAP inactivating kinase Lats1/2 [75,77]. Notably, also F-actin polymerization status and change of cell adhesion mediated by Rho GTPase deactivation and cytoskeletal reorganization were shown to be correlated with YAP activation [75,77]. In general, from these studies, it emerges that increased Rho GTPase activity and actin polymerization activate, whereas destabilization of actin inhibit, YAP and TAZ [77].
High basal and post-PH levels of YAP are counteracted by high levels of YAP inactivating kinases in aged livers
As to the role of YAP during liver regeneration post-PH, Pibiri et al. [45] found that while YAP protein expression essentially reflected progression into cell cycle in young mice, an early, robust and persistent increase of the protein occurred in aged mice. In spite of YAP increase, liver regeneration resulted reduced in aged animals. According to the Authors, the persistent increased levels of YAP in aged liver represents a signal that senses the decreased liver mass, and consequently, it is aimed at stimulating a regenerative response in order to re-establish the original liver size [45]. More recently, Laforese et al. [68] found that aged mouse livers had higher basal levels of YAP than young ones. Interestingly, aged animals also showed higher basal and post PH levels of the active form of the YAP inactivating kinases MST and LATS [68] compared to young mice. Furthermore, siRNA-mediated silencing of MST1 and MST2 was able to induce hepatocyte proliferation in quiescent young livers and to rescue regeneration in aged ones following PH [68]. These findings suggest a direct link between YAP activation and liver regeneration. Nevertheless, the involvement of MST1 in diverse signaling pathways [78–80], does not rule out the possibility that cooperation of YAP with other effectors is also involved in liver regeneration.
Cooperation between YAP and BubR1 in mediating liver regeneration: a possible scenario
Given the findings that: i) BubR1 is a direct transcriptional target of YAP [57]; ii) both YAP and BubR1 are involved in regulating cell adhesion and positively modulate cell proliferation [46,57,61,63,64,67]; iii) both BubR1 and YAP expression/activation are reduced in condition of impaired liver regeneration [45,57,68], the following scenario can be hypothesized. Under physiological condition the Hippo pathway efficiently controls the size of the liver. Indeed, cell-cell and cell-extracellular matrix contacts and high cell density activate the Hippo pathway through phosphorylation-dependent inactivation of YAP and its retention within the cytoplasm. In young mice, reduction of the liver mass after PH leads to YAP activation and subsequent transcription of BubR1 which, in turn, induces DISC1 protein, thus guaranteeing a proper micro structural adaptation during tissue regeneration. In aged livers, however, the increased basal levels of YAP inhibitory kinases, such as MSTs, counteract the positive signals consequent to loss of liver mass [68]. As a result, despite increased levels of YAP proteins, in aged hepatectomized livers the tissue mass cannot be properly restored due to the YAP inactivating phosphorylation mediated by MSTs [68]. Consequently, the decreased transcription of BubR1, a YAP target gene, leads to DSC1 reduction, weakened micro structural adaptation and p21 upregulation which, in turn, causes G1 cell cycle arrest and impaired liver regeneration (Fig 1). This scenario implies a coordinate action between YAP and BubR1 to rescue conditions at risk of tissue damage, such as liver regeneration after surgery or the worsening of necrosis induced by cytotoxic agents. It follows that uncoupling of their activity may impair liver regeneration in aged liver, but only in association with tissue loss. According to this view, one may explain why aged hepatocytes still retain their fully replicative capacity following exposure to direct mitogenic stimuli, which are able to induce hepatocyte proliferation in the absence of tissue damage [81].
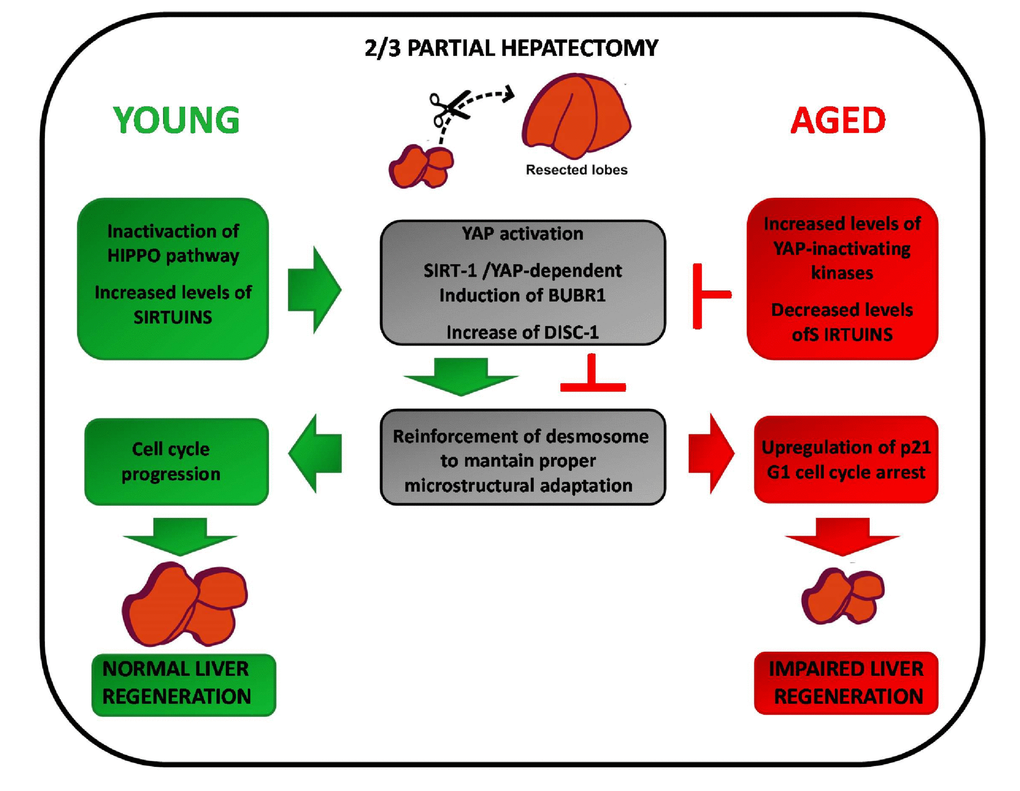
Figure 1. Intracellular factors affecting compensatory regeneration in aged livers.Young livers (left): reduction of the liver mass after PH leads to YAP/Sirtuin-dependent transcription of BubR1. BubR1, in turn, induces the cell adhesion protein DISC1 which provides the proper microstructural adaptation during regeneration. Aged livers (right): the increased levels of MSTs counteract YAP activation. Furthermore, as aging is associated to decreased SIRT-1 expression, there is a reduction of the YAP/Sirtuin-dependent transcription of BubR1. This lead to reduction of DSC1 expression, weakened microstructural adaptation and upregulation of p21. As a result, G1 cell cycle arrest and impairment of liver regeneration is observed.
Role of factors affecting the circadian clock: SIRT1
The circadian clock
The cellular and physiological rhythms observed over a 24 hour period are termed circadian rhythms. These regulate many physiological processes, such as sleep/awake, metabolism and hormonal secretion [82,83]. In mammals, the circadian timing system is composed of a central pacemaker, housed in the brain’s suprachiasmatic nucleus (SCN). The rhythm-generating mechanism is thought to rely on a feedback loop involving positively and negatively acting transcription factors [84–86]. More in detail, aryl hydrocarbon receptor nuclear-translocator-like (ARNTL or BMAL1) and Circadian Locomotor Output Cycles Kaput (CLOCK) transcription factors activate the expression of Period (Per) and Cryptochrome (Cry) genes, and once PER and CRY repressor proteins accumulate to a critical level they bind to BMAL1-CLOCK heterodimers and thereby repress the transcription of their own genes [84–86]. Although the master circadian clock is located within the SCN, the key circadian proteins are expressed in many peripheral tissues, determining circadian periodicity in gene expression and physiology for many organs [86]. Indeed, microarray data have shown that up to 10% of genes in different tissues are directly or indirectly regulated by the circadian clock system [87,88].
Aging is associated to degradation of the neuronal activity rhythms in SCN and to deregulated nutrient sensing
It is commonly assumed that the circadian clock and the aging process are intimately entangled. More specifically, it has been demonstrated that changes in central rhythmic behavior occur with ageing [89], mainly due to the age-dependent degradation of the neuronal activity rhythms in SCN [90]. On the other hand, mice deficient in core circadian transcription factor (BMAL1) expression have reduced lifespan and premature aging phenotype [91]. Aging-associated deterioration is associated to disruption of metabolic homeostasis, including deregulated nutrient sensing [92–97]. Accordingly, caloric restriction (CR) has been found to extend lifespan in several organisms and to rewire circadian metabolism [97–102].
Sirtuins
Sirtuins are a nicotinamide adenine dinucleotide (NAD+)-dependent family of histone deacetylases (HDACs) which are involved in various physiological functions, such as aging, genome integrity maintenance, stress response to nutrient challenge, metabolic control and cancer [103–105]. HDACs mediate deacetylation of histones which leads to gene silencing [106–108] but are also implicated in the reversible acetylation of non histone proteins [109–111]. The catalytic reaction mediated by sirtuins involves the breakdown of one molecule of NAD+ for each deacetylated acetyl lysine and the generation of nicotinamide and O-acetyl-ADP-ribose, which serves as an acyl acceptor to form an acylADP-ribose product. Similar to other posttranslational modifications, the presence or absence of acyl groups on specific lysine residues in proteins can determine their function and subcellular localization [112–114]. Apart from their deacetylase function, the seven mammalian members of the sirtuin family have different enzymatic activity, biological targets and cellular functions [115,116].
SIRT-1 contributes to the modulation of the circadian clock by nutrients and of the aging process
SIRT-1, member of sirtuin family, has been found to deacetylate histones and several transcription factors involved in the control of metabolism [117–121], in particular following nutritional deprivation [121–123]. Recently, Orozco-Solis et al. analyzed the role played by SIRT-1 in the modulation of the SCN by nutritional inputs by using mice with Sirt1 ablation in the steroidogenic factor 1(Sf1) neurons of the ventromedial hypothalamus (VMH) [124]. Indeed, Sf1-expressing neurons, exclusively located in the VMH, were demonstrated to be required for the regulation of energy balance and glucose metabolism [125,126]. They found that SIRT-1 in the VMH operated as a metabolic sensor able to connect food intake to circadian behavior. Indeed, under food restriction and absence of light, SIRT-1 contributed to activity behavior which was associated to changes in the acrophase and amplitude of core clock genes in the SCN. Thus, under specific physiological conditions, SIRT1 contributes to the modulation of the circadian clock by nutrients.
Recent studies have also documented a link between SIRT-1-dependent core clock gene regulation and aging [114,127]. Accordingly, it has been demonstrated that the brain-specific Sirt1−/− (BSKO) mice exhibited dampened circadian gene expression in the anterior hypothalamus (where the SCN is located). This was associated to a lengthened circadian period and an accelerated aged phenotype [128]. Thus, loss of SIRT-1 in the brain not only regulates the circadian clock but also accelerates the aging process, which is most likely mediated by NAD+. The additional findings of the mammalian SIRT-1 involvement in circadian control of liver gene expression and metabolism has documented the existence of an interplay between aging, nutritional challenge and circadian metabolism [103,129,130].
Caloric restriction is able to rescue the decline in rhythmic global protein acetylation of aged liver by enhanced activity of SIRT-1
Recently, Sato et al. [131] analyzed the mechanisms associated to CR-dependent improvement of circadian rhythms. To this aim, they have paralleled the transcriptomic profile of liver cells with those of muscle and epidermal stem cells of young and old mice [132] [104], fed ad libitum with either normal chow diet or under caloric restriction (CR). Data obtained revealed that aging and CR caused remarkable tissue-specific circadian changes in the liver and led to the identification of a rhythmic global protein acetylation as a hallmark of the liver clock. Remarkably, this oscillatory acetylation signature was drastically affected in old mice and was rescued under CR regimen. The authors attributed changes in CR-associated protein acetylation to improvement in NAD+ availability, increased levels of acetate and acetyl-CoA and enhanced activity of SIRT1 [133–135]. Accordingly, an enrichment of SIRT1-target genes was found in mouse livers under CR [136]. Control of protein acetylation was shown to be mediated at least in part, by SIRT1 and the dynamic acetylation of cytoplasmic Acetyl-CoA Synthetase 1 (AceCS1). Indeed, previous studies demonstrated that SIRT-1 was able to deacetylate and control cytoplasmic AceCS1 [137], as a regulatory circadian pathway which results in the production of cyclic pools of acetate-derived acetyl-CoA [138]. Accordingly, CR was associated to increased hepatic acetate levels in young mice and extended acetylation profile of AceCS1 in young and old animals [131]. Although a causal relationship was not directly demonstrated, Authors have hypothesized that the change in circadian acetylation of AceCS1 could be linked to the increased levels of acetyl-CoA and global protein hyperacetylation observed under CR [131]. Summarizing, this study demonstrates that aging affects cyclic global protein acetylation, mediated by timely activation of both histone acetyltransferases (HATs) and HDACs, which can be rescued by nutritional deprivation.
Decreased SIRT-1 expression in aged liver may impair liver regeneration via reduction of BubR1
A direct molecular link between the circadian clock and cellular proliferation has been established [139]. Indeed, rhythmic expression of key regulators of cell cycle progression and of the DNA-damage response is dependent on the circadian machinery [140–142]. Moreover, liver regeneration is impaired in mice deficient in core clock genes [143]. Based on these findings and on the role of SIRT-1 in regulating circadian genes, Bellet et al., analyzed liver regeneration following PH in liver-specific Sirt1-deficient mice [144]. Their results showed that G1/S progression, as well as circadian gene expression, were significantly affected by the loss of SIRT-1.
These results suggest that the decreased expression of SIRT-1 observed in aged liver [131] could impair liver regeneration post PH. In this context, notably, Singh et al. identified BUB family proteins, i.e.BubR1, as downstream targets of SIRT-1 in melanoma cells [145]. In particular, they demonstrated that specific inhibition of SIRT-1 by tenovin-1 was associated to decrease of both protein and mRNA levels of BuB proteins. Thus, not only decreased YAP activation, but also decreased SIRT-1 levels could cooperate to determine BubR1 reduced expression in aged livers, thereby affecting liver regeneration after tissue injury [68,144,145]. (Fiure 1).
Extra-cellular factors affecting liver regeneration in aged rodents
It was shown that aging does not affect mouse hepatocytes replicative capacity following exposure to mitogenic agents, such as nuclear receptor ligands (NRL) [81], able to induce cell proliferation in the absence of tissue injury (direct hyperplasia). The early signal transduction pathways involved in nuclear receptor-mediated hepatocyte proliferation are quite different from those of liver regeneration. Indeed, neither activation of transcription factors (AP-1, NF-κB, STAT3, and C/EBP), nor increased expression of immediate early/growth factor genes (c-Fos, c-Jun, c-Myc, Lrf-1, Egr1, Hgf and Tgfα) or release of the cytokines tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6) could be observed in rodents after treatment with NRL [146–151]. Those results demonstrate that the intrinsic hepatocyte replicative capacity is maintained during aging if an appropriate proliferative stimulus is provided. Based on this, it has been suggested that extrinsic factors (i.e. growth factors, cytokines, hormones) rather than intrinsic changes within the cell could be responsible for the depressed replicative response observed in aged rodent liver [81]. Accordingly, a pioneer study of Conboy at al [152], reported that heterochronic parabiosis (a shared circulatory system between young and old mice) was able to increase proliferation of hepatic progenitor cells of aged animals. Enhanced liver cell proliferation was associated with a decrease in aged livers of the c/EBPα complex, a known cell proliferation inhibitor whose expression increases with age. This finding suggested the existence of systemic factors able to modulate the molecular signaling pathways critical to liver-specific progenitor cell activation which are present in young liver microenvironment but are lost in aged livers.
A number of extra-cellular factors has been implicated in age-dependent impairment of liver regeneration, including growth hormone (GH) [153], Src homology 2 domain-containing (Shc) protein p66Shc [154] and interferon gamma (IFN-ϒ) [155].
As to GH, Moolten et al. showed that its administration to rats accelerated liver regeneration after PH [156]. Later studies by Krupczak et al. [153] found that GH administration to old mice was able to improve liver regeneration post-PH by increasing FoxM1b levels to those seen in young animals after surgery. Moreover, Jin et al. [157] demonstrated that GH improved liver regeneration through glycogen synthase kinase 3 (GSK3) regulation. In particular, they showed that in young animals, high levels of GH increased the expression of GSK3. This kinase associated to and degraded cyclin D3, an activator of cdk4 which positively regulates the growth inhibitor C/EBPα in aged mouse livers [158]. Opposite, in aged livers the GSK3 promoter was found to be repressed by C/EBP-histone deacetylase 1 (HDAC1) complexes, leading to the GSK3 reduction. Accordingly, treatment of old mice with GH increased expression of GSK3 via removal of the C/EBP-HDAC1 complexes from the GSK3 promoter. Furthermore, while down-regulation of GSK3 in young mice inhibited liver cell proliferation post-PH, via the cyclin D3-C/EBP pathway, its up-regulation in old mice accelerated hepatocyte proliferation [157]. On this basis, GSK3 has been considered as a key regulator of liver cell proliferation whose age-dependent reduction affected the liver regenerative capacities.
p66Shc protein is a member of the Src homology 2 domain-containing (Shc) A family, ubiquitously expressed except for in the brain and in neurons [154]. ShcA proteins have been found to act as adaptor molecules involved in epidermal growth factor-mediated Ras/mitogen-activated protein kinase (MAPK) cascade activation. However, p66Shc was shown to inhibit activation of the Ras/MAP kinase pathway, by competing with p46Shc and p52Shc for binding to the adaptor protein Growth factor receptor-bound protein 2 (Grb2) [154,159–162]. Moreover, it was shown to induce cell oxidative stress (OS) and apoptosis [163–165]. Accordingly, loss of p66Shc has been shown to be associated to increased resistance to OS and apoptosis [163,164] and to protection against ROS-mediated acute tissue damage [165]. Furthermore, although aging is characterized by elevated levels of ROS, p66Shc knockout mice display a higher resistance to OS and an increased lifespan compared to their wild-type counterpart [166]. Thus, p66shc has been proposed to act by monitoring the intracellular concentration of ROS in order to eliminate cells injured by oxidative damage [167]. On these premises, Haga et al. [167] decided to analyze the role of p66Shc in liver regeneration post-PH during ageing. They found that impairment of tissue recovery in aged mice was associated to OS, which was an early event post-surgery, followed by marked apoptosis. Interestingly, p66Shc was strongly phosphorylated at S36 early after PH. Notably, this phosphorylated form of p66Shc (p6636S) has been reported to induce cellular OS by suppressing the activity of the anti-oxidant/pro-survival molecule Akt and the expression of catalase [163,164,166]. Furthermore, p6636S seems to be critical for induction of apoptosis in cell exposed to OS [163,164,166]. Accordingly, while several markers of cell proliferation were expressed/activated after PH in livers of both aged and young mice, Akt was not activated in the former group [167]. Ablation of hepatic p66Shc remarkably improved liver regeneration post-surgery in aged animals, as well as Akt activation, which was associated to suppression of post-surgery OS and apoptosis. Interestingly, loss of p66Shc did not affect post-PH liver regeneration in young mice. Thus, these findings [168] suggest a key role for p66Shc in the impairment of liver regeneration during aging.
Several studies have shown an up-regulation of the inflammatory response with age across various tissues [168,169]. Accordingly, Singh et al. [170] described immune cell infiltration and elevation of the transcript levels of various growth inhibitory inflammatory cytokines, such as transforming growth factor-β (TGF-β), IFN-ϒ and interleukin (IL)-1, in the liver of aged animals.
IFN-ϒ inhibits cell cycle through activation of the cyclin-cdk complex inhibitor p21 via phosphorylation of the STAT1 protein, which, in turn, activates the IFN-specific transcription factor IRF1 [171]. IRF1 causes p53 nuclear accumulation [172,173] and increased nitric oxide production via inducible nitric oxide synthase induction [174,175] which, subsequently, lead to p21 transcription [176]. Based on these findings, Singh et al. analyzed the role of IFN-ϒ in the impairment of liver regeneration post-PH in aged mice [177]. They found higher IFN-ϒ levels in aged livers compared to young tissues which were associated to increased IFN-ϒ-dependent gene transcription both before and after PH. The increased IFN-ϒ levels observed in aged livers negatively correlated with liver regeneration post-surgery. In vivo deletion of macrophages and natural killer cells, the major IFN-ϒ producers, led to restoration of the proliferation kinetics post-PH in aged mice. Furthermore, aged IFN-ϒ−/− mouse livers exhibited an earlier entry into the cell cycle compared with age-matched controls. Thus, these findings strongly suggest that the age-related increase in the pro-inflammatory status in the liver [170] has a detrimental effect on its regenerative capacity. From these studies it emerges that the age-dependent increase of oxidative stress and inflammatory molecules plays a crucial role in liver regeneration.
Extracellular factors involved in the regulation of oxidative and inflammatory status also represent the focus of the most recent studies on liver regeneration during ageing (Figure 2).
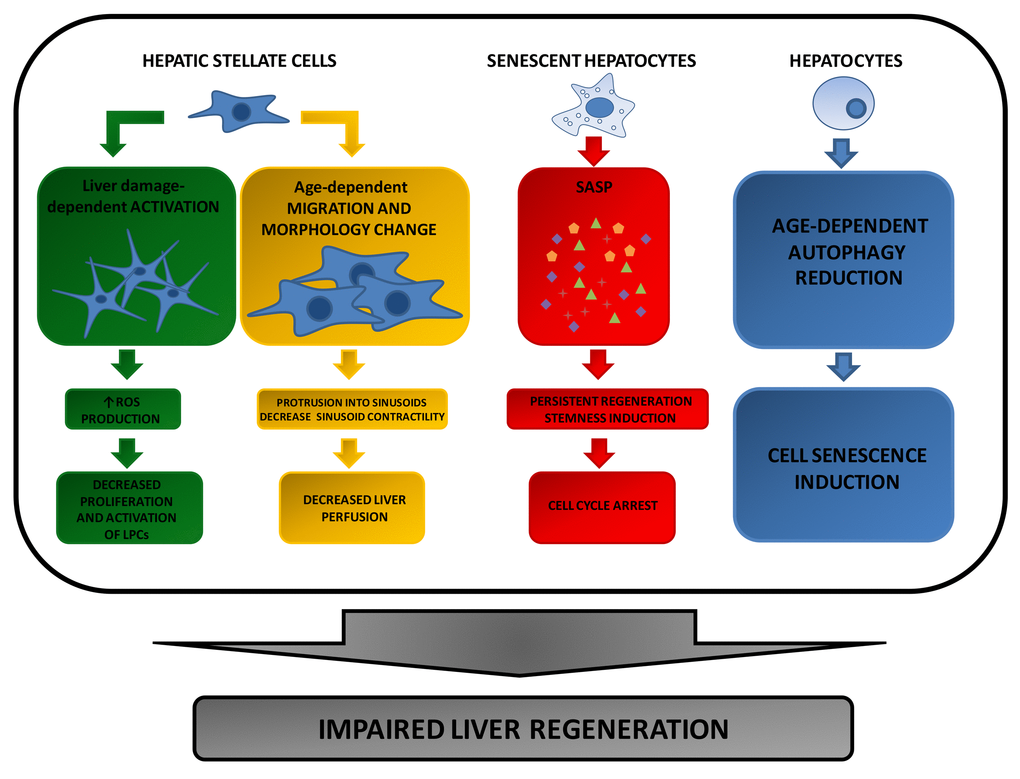
Figure 2. Extracellular factors affecting compensatory regeneration in aged livers. During ageing, activated HSC-induced chemokine production leads to a decreased activation and proliferation of Liver Progenitor Cells (LPCs). Since these cells are required to repopulate the liver following tissue injury, their decrease results in a reduced liver regenerative response. Furthermore, age-dependent HSC size increase and migration, can negatively affect hepatocyte proliferation by reducing liver perfusion. A further reason for the impaired liver regeneration of aged hepatocytes is due to the pro-inflammatory proteins chronically released by senescent hepatocytes that accumulate in the elderly as a consequence of a reduction of autophagy program.
Role of factors affecting oxidative status and liver perfusion: changes in hepatic stellate cell (HSC) activity, phenotype and localization
HSCs mediate the negative regulation of LPCs during liver regeneration in aged animals
Based on the free radical theory, ageing is the consequence of the accumulation of reactive oxygen species (ROS) due to free radical incomplete destruction by the appropriate endogenous defense systems [178]. While generation of ROS is essential to maintain cellular homeostasis, when excessive, it might lead to tissue damage and to activation of specific signaling pathways influencing aging and age-related disease development [179].
Recently, Cheng et al. [180] have demonstrated that oxidative stress inhibits liver progenitor cell (LPC) activation in aged mice during liver regeneration. In particular, they [180] have analyzed liver regeneration following choline devoid-ethionine supplemented (CDE) diet. This dietary regimen causes liver damage and expansion of LPCs, also known as ‘oval cells’ [181]. Upon massive liver injury, LPCs proliferate and migrate into the hepatic lobule where they can differentiate into hepatocytes and/or biliary epithelial cells [182–186]. Notably, LPC expansion occurs in many human liver diseases [187] and experimental animal models [188].
In the study of Cheng et al. [180], it was shown that CDE administration induced LPC proliferation in young mice, but not in old animals. Loss of LPC proliferation was associated to the impairment of the regenerative response of aged livers. Interestingly, no significant difference in the clonogenic and proliferative capacity of isolated LPCs was found between young and old mice. Thus, no intrinsic cellular changes can be claimed as causative factors for age-dependent reduction in liver regeneration. Accordingly, the age-dependent decrease of LPC proliferation appeared to be due to extracellular factors, in particular excessive ROS levels produced by neutrophils recruited into the cell niche by activated hepatic stellate cells (HSCs) [180] .
Summarizing, in this study HSCs are recognized as the critical cell population in the negative regulation of LPCs during liver regeneration in aged animals. Nevertheless, more recent studies questioned the role of LPCs in repopulating the liver after tissue damage [189–193]. Indeed, novel genetic lineage tracing experiments have shown that virtually all hepatocytes in the injured liver derive from self-expansion of the pre-existing hepatocytes [185,186,194,195]. These studies attributing to LPCs a negligible contribution in liver regeneration cast doubts about a critical role of HSCs in liver mass recovery. Nevertheless, HCSs have been suggested to affect liver regeneration during ageing by further mechanisms.
Changes in HSC phenotype and localization during aging could affect liver regeneration response via reduction of tissue perfusion
An additional role of HSCs in the ageing process that could be related to the impairment of the liver regenerative capacity has been proposed by Marcos et al. [196]. These Authors found that during aging, HSCs migrate from the centrilobular to the periportal areas where they display a modified morphology characterized by cell enlargement due to increased content of lipid droplets. In particular, during ageing HSCs morphology changes from a small cell body with long and thin extensions to a large cell body with thicker and much shorter extensions. A larger cell body of HSCs has been implicated in the blood flow reduction observed in the sinusoids of older animals [197], due to cell protrusion into sinusoids [198]. Marcos et al. suggested that also the age-dependent shortening of HSC processes could play a role in liver flow reduction [196]. Indeed, the HSC processes have been shown to exert a pivotal role in sinusoid contractility acting as chemotactic signal sensors [199]. Accordingly, it has been hypothesized that the shorter processes observed in aged HSCs could surround and control the blood flow of fewer sinusoids than in young mice where HSCs encircled more than two sinusoids [200]. Even if the study of Marcos et al. [196] was not focused on liver regeneration, it provides another possible explanation for the impaired liver regeneration observed in the elderly, as already suggested by Saito et al. [201].
Role of factors affecting inflammation status: SASP
Cellular senescence and “senescence-associated secretory phenotype” (SASP)
Chronic inflammation is associated to normal aging and to several age-related diseases, such as cancer, atherosclerosis and osteoarthritis [202,203]. While in the past it has been mainly attributed to the progressive activation of immune cells over time [202,204,205], recent studies have proposed that cellular senescence might be an important additional contributor to chronic inflammation [206–208].
Cellular senescence is thought to be a tumor suppressive stress response [209–211] which is also associated with aging. Indeed, senescent cells accumulate with ageing and a variable proportion of them, interspersed in islands of normal cells, is a common feature in aging mammalian tissues [212–215].
Wang et al. [212] have examined the process of hepatocyte senescence in normal mouse liver during a time period of 18 months. They have found that the percentage of hepatocytes characterized by polyploidy, accumulation of DNA-damage and activation of p21 and p16ink4, all common features of cell senescence, increased with age [212]. This accumulation of cell cycle arrested cells leads to the age-dependent loss of functional and regenerative tissue capacity which prevents proliferation of somatic and stem cells in a cell intrinsic-manner [216–222]. Common age-dependent cell intrinsic defects comprehend p53 activation and increased expression of the cyclin-dependent kinase inhibitor p16 INK4a [216–222].
Similar to mouse hepatocytes, human hepatocytes undergo age-dependent senescence [212,223] and it has been shown that poor liver regeneration in older patients was associated with up regulation of senescence-related genes, such as p16, and down-regulation of regeneration-promoting ones, such as HGF and MET [224].
Senescent cells have been demonstrated to disrupt tissue structure and function through secretion of pro-inflammatory factors participating in intercellular signaling [206–208,225]. Indeed, despite of an apparently irreversible growth arrest, these cells are metabolically active and are characterized by a peculiar morphology, physiology and gene expression [225]. Secretion of pro-inflammatory factors has been termed as “senescence-associated secretory phenotype”, or SASP, and it is generally induced at transcriptional level [206]. SASP includes a wide range of growth factors, proteases, chemokines and cytokines (i.e. pro-inflammatory proteins IL-6, IL-8, IL-1, granulocyte macrophage colony stimulating factor (GM-CSF), growth regulated oncogene (GRO)α, monocyte chemotactic protein (MCP)-2, MCP-3, MMP-1, MMP-3, and many of the Insulin-like growth factor (IGF)-binding proteins) [206,226–228]. In addition, to reinforce the growth arrest and induce senescence in a paracrine manner [222,225,229], SASP has also been shown to favor wound healing [230], embryonic development [231,232] and tumor growth [233,234], suggesting more complex physiological roles than currently understood.
SASP could affect liver regeneration during aging by blocking neighboring cells in a stem like state
A possible explanation to the apparent discrepancy between the growth inhibitory and tumor suppressive effect of senescence and the tumor promotion activity of SASP, could be found in the recent study of Ritschka et al. [235]. Briefly, these Authors have shown that transient exposure of mouse primary keratinocytes to SASP determined increased expression of stem cell markers and regenerative capacity in vivo. On the opposite, a prolonged exposure caused a subsequent increased stemness which was blocked by a cell-intrinsic senescence arrest mediated by the cell cycle inhibitor p19. These results were interpreted to suggest that cells sensed a prolonged SASP-induced regeneration as a pro-tumorigenic event, and, therefore, activated cell-intrinsic tumor-suppressive mechanisms as a protection devise. Furthermore, in the same study, it was demonstrated that senescence induction in single liver cells in vivo was associated to activation of tissue-specific expression of stem cell markers. This has supported the finding that SASP can induce stemness in the surrounding tissue in a paracrine manner [235]. According to the Authors, the discovery that timely controlled exposure to SASP can directly promote tissue regeneration identifies new important biological roles for the senescence program, supporting the idea that senescence might be primarily a beneficial and pro-regenerative process. However, when perturbed, it can result in tumor formation and aging.
More recently, de Keizer et al. [236] have hypothesized that the SASP ability to trigger reprogramming of neighboring cells into more pluripotent cells in vivo [235,237] could be responsible for the reduced tissue regeneration capacity during aging. Indeed, they have proposed that in young tissues the presence of few senescent cells would permit the turnover of damaged or lost cells by a transient SASP response. This, could lead to temporary cell reprogramming and subsequent proliferation/differentiation. Opposite, the chronic release of SASP factors in aged tissue, due to senescent cell accumulation, would effectively make this cell reprogramming permanent. As a consequence, this would keep the neighboring recipient cells locked in this stem-like state, affecting replacement of lost cells in aged tissue [236].
Tissues of BubR1 deficient mice are characterized by senescent hepatocytes/SASP increase
A further indication of the possible involvement of SASP in the impairment of tissue regeneration in elderly, stems from a previous study of Baker et al. [50]. In this study it was shown accumulation of p16INK4a positive senescent cells and increased SASP in fat and muscle tissues of BubR1 deficient mice [50] which develop early ageing-associated phenotypes [49–53]. In the same study, deletion of p16 coding gene was found to be associated to increased cell replication and decreased SASP. Furthermore, a later study found that clearance of senescent cells in tissues of BubR1 insufficient mice was able to prevent or delay tissue dysfunction and to extend health span [238]. Since BubR1 deficiency is involved in age-dependent impaired liver regeneration [25], collectively, these findings suggest a possible role for SASP in the reduced liver regenerative response.
Autophagy reduction, cell senescence and impairment of regeneration in aged livers
The major inducible pathways for degradation of cellular constituents in eukaryotic cells is represented by autophagy. Autophagy plays an essential role in cellular metabolism and homeostasis by degrading both long-lived cytoplasmic proteins and dysfunctional organelles via lysosome-dependent machinery [239,240]. Recently, the potential involvement of autophagy in aging and aging-associated organ injuries has become evident. Accordingly, to Baker’s study [238], defective autophagy reduces the lifespan, whereas induction of autophagy has been associated to increased longevity in multiple animal species [241,242]. Furthermore, it has been demonstrated that aged livers are characterized by a decline in autophagy activity and that its restoration is able to improve cellular maintenance and hepatic function [243,244].
Recently, Liu et al. [245] showed that plasma from young mice attenuates hepatic injury and restores liver regeneration capacity after PH in aged animals. Indeed, reduction of endoplasmic reticulum stress, hepatocellular damage and autophagosomes and an increase of proliferating cells were observed in aged livers treated with young plasma. Intriguingly, this effect was abolished under administration of autophagy inhibitors. This finding has been further consolidated by in vitro data showing that young serum prevented old hepatocytes from senescence and that its effect was abrogated by autophagy inhibition [245]. Based on these observations, the Authors concluded that restoration of autophagy could inhibit cellular senescence supporting a direct link between senescence, autophagy and liver regeneration. Indeed, since senescent cells alter tissue structure and function through SASP release, their elimination by autophagy could alleviate aging-induced hepatic injury and restore the regenerative response.
Conclusion
The long standing concept that hepatocytes lose their proliferative capacity with ageing has been challenged by several experimental evidences based on a successful expansion of hepatocyes even after several rounds of transplantation [212]. Remarkably, aged hepatocytes also retain their fully proliferative capacity if exposed to the treatment with direct mitogenic stimuli, such as ligands of the nuclear receptor CAR, which do not cause liver injury [81].
More recently, increasing evidence suggests that the age-dependent decline of the liver regeneration capacity is the consequence of multiple intertwining factors, both intra and extra-cellular, that cooperate to affect liver mass recovery after tissue damage. From the analysis of the latest literature reports, it emerges that the mass recovery of the injured liver in aged animals is compromised by at least three factors:
i) decreased expression of cell adhesion proteins leading to weakened microstructural adaptation after tissue injury and p21-dependent cell cycle arrest [46];
ii) change of HSC morphology which results in reduced liver perfusion and, consequently, leads to an impairment of tissue reconstitution after damage [196];
iii) chronic release of stemness-inducing pro-inflammatory proteins by senescent hepatocytes, which accumulate in the elderly due to a decline of the autophagy program [235,245].
Thus, while no major age-dependent changes in the expression/activation of factors classically associated to liver regeneration (IEGs, genes coding for cytokines/growth factors or transcription factors) have been convincingly demonstrated [45], it is becoming evident that the altered expression/function of genes (BubR1, YAP, SIRT-1) and cells (HSCs) directly or indirectly involved in injured tissue reconstitution are responsible for the impairment of liver regeneration during ageing [46,57,144,145,196]. Such an hypothesis also explains why no difference in the proliferative capacity of aged and young hepatocytes can be observed following administration of mitogenic agents devoid of hepatotoxicity [81].
An additional cause of the age-dependent decline of hepatocyte proliferation has been identified in the senescent hepatocyte-induced SASP release. Indeed, SASP accumulate during ageing consequently to a decline of the autophagy program; SASP accumulation, in turn, maintains the neighboring recipient cells locked in a stem like state in aged tissues, affecting their capacity to replace lost cells [236].
Age-dependent decreased expression of genes regulating cell adhesion as well as SASP secretion by senescent cells might also be dictated by changes in circadian rhythms occurring during aging [131,145,236], adding further complexity to this topic.
On these bases, a potential therapeutic approach of direct mitogens to relieve the proliferative decline taking place in aged injured liver could be proposed [24]. Treatment with nuclear receptor ligands could also be useful in liver transplantation and hepatic failure in order to restore liver function. Furthermore, therapeutic interventions aimed at eliminating senescent cells or blocking their effects may be useful to treat or delay age-related diseases [236]. In this regard, it also would be interesting to evaluate if ligands of nuclear receptors could have a role in this process. Indeed, as nuclear receptors are ligand-induced transcription factors [246], their activation could unlock SASP-mediated senescence-stem locked cells by reprogramming their gene expression thus eliciting a similar hepatocyte proliferation response in young and aged livers.
In conclusion, in view of the many still unanswered questions, a greater understanding of the molecular mechanisms responsible for the impairment of the regenerative response of the liver is both a priority and a fascinating scientific challenge that may promote the development of innovative concepts that can be translated into the clinic.
Conflicts of Interest
The author of this review has no conflicts of interest to disclose.
Funding
The author has no funding to acknowledge.
References
- 1. Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006 (Suppl 1); 43:S45–53. https://doi.org/10.1002/hep.20969 [PubMed]
- 2. Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007; 213:286–300. https://doi.org/10.1002/jcp.21172 [PubMed]
- 3. Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997; 276:60–66. https://doi.org/10.1126/science.276.5309.60 [PubMed]
- 4. Diehl AM, Rai R. Review: regulation of liver regeneration by pro-inflammatory cytokines. J Gastroenterol Hepatol. 1996; 11:466–70. https://doi.org/10.1111/j.1440-1746.1996.tb00292.x [PubMed]
- 5. Higgins GM, Anderson RM. Experimental pathology of the liver. Restoration of the liver of the white rat following partial removal. Arch Pathol (Chic). 1931; 12:186–202.
- 6. Michalopoulos GK. Hepatostat: liver regeneration and normal liver tissue maintenance. Hepatology. 2017; 65:1384–92. https://doi.org/10.1002/hep.28988 [PubMed]
- 7. Mars WM, Zarnegar R, Michalopoulos GK. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol. 1993; 143:949–58. [PubMed]
- 8. Mars WM, Liu ML, Kitson RP, Goldfarb RH, Gabauer MK, Michalopoulos GK. Immediate early detection of urokinase receptor after partial hepatectomy and its implications for initiation of liver regeneration. Hepatology. 1995; 21:1695–701. [PubMed]
- 9. Lindroos PM, Zarnegar R, Michalopoulos GK. Hepatocyte growth factor (hepatopoietin A) rapidly increases in plasma before DNA synthesis and liver regeneration stimulated by partial hepatectomy and carbon tetrachloride administration. Hepatology. 1991; 13:743–50. https://doi.org/10.1002/hep.1840130422 [PubMed]
- 10. Stolz DB, Mars WM, Petersen BE, Kim TH, Michalopoulos GK. Growth factor signal transduction immediately after two-thirds partial hepatectomy in the rat. Cancer Res. 1999; 59:3954–60. [PubMed]
- 11. Paranjpe S, Bowen WC, Mars WM, Orr A, Haynes MM, DeFrances MC, Liu S, Tseng GC, Tsagianni A, Michalopoulos GK. Combined systemic elimination of MET and EGFR signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology. 2016; 64:1711–24. https://doi.org/10.1002/hep.28721 [PubMed]
- 12. Michalopoulos GK. Principles of liver regeneration and growth homeostasis. Compr Physiol. 2013; 3:485–513. https://doi.org/10.1002/cphy.c120014 [PubMed]
- 13. Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004; 5:836–47. https://doi.org/10.1038/nrm1489 [PubMed]
- 14. Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996; 274:1379–83. https://doi.org/10.1126/science.274.5291.1379 [PubMed]
- 15. Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA. 1997; 94:1441–46. https://doi.org/10.1073/pnas.94.4.1441 [PubMed]
- 16. Bhave VS, Paranjpe S, Bowen WC, Donthamsetty S, Bell AW, Khillan JS, Michalopoulos GK. Genes inducing iPS phenotype play a role in hepatocyte survival and proliferation in vitro and liver regeneration in vivo. Hepatology. 2011; 54:1360–70. https://doi.org/10.1002/hep.24507 [PubMed]
- 17. Fausto N, Webber EM. Liver regeneration. In: Arias IM, Boyer JL, Fausto N, Jakoby WB, Schachter DA, Shafritz DA. 1994 eds. The liver: biology and pathobiology. Ed 3. New York: Raven, 1059-1084.
- 18. Marie Scearce L, Lee J, Naji L, Greenbaum L, Cressman DE, Taub R. Rapid activation of latent transcription factor complexes reflects initiating signals in liver regeneration. Cell Death Differ. 1996; 3:47–55. [PubMed]
- 19. Greenbaum LE, Li W, Cressman DE, Peng Y, Ciliberto G, Poli V, Taub R. CCAAT enhancer- binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J Clin Invest. 1998; 102:996–1007. https://doi.org/10.1172/JCI3135 [PubMed]
- 20. Song G, Sharma AD, Roll GR, Ng R, Lee AY, Blelloch RH, Frandsen NM, Willenbring H. MicroRNAs control hepatocyte proliferation during liver regeneration. Hepatology. 2010; 51:1735–43. https://doi.org/10.1002/hep.23547 [PubMed]
- 21. Ng R, Song G, Roll GR, Frandsen NM, Willenbring H. A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J Clin Invest. 2012; 122:1097–108. https://doi.org/10.1172/JCI46039 [PubMed]
- 22. Van Thiel DH, Gavaler JS, Kam I, Francavilla A, Polimeno L, Schade RR, Smith J, Diven W, Penkrot RJ, Starzl TE. Rapid growth of an intact human liver transplanted into a recipient larger than the donor. Gastroenterology. 1987; 93:1414–19. https://doi.org/10.1016/0016-5085(87)90274-5 [PubMed]
- 23. Zhu C, Ikemoto T, Utsunomiya T, Yamada S, Morine Y, Imura S, Arakawa Y, Takasu C, Ishikawa D, Shimada M. Senescence-related genes possibly responsible for poor liver regeneration after hepatectomy in elderly patients. J Gastroenterol Hepatol. 2014; 29:1102–08. https://doi.org/10.1111/jgh.12468 [PubMed]
- 24. Bucher NL, Swaffield MN, Ditroia JF. The influence of age upon the incorporation of thymidine-2C14 into the DNA of regenerating rat liver. Cancer Res. 1964; 24:509–12. [PubMed]
- 25. Fry M, Silber J, Loeb LA, Martin GM. Delayed and reduced cell replication and diminishing levels of DNA polymerase-alpha in regenerating liver of aging mice. J Cell Physiol. 1984; 118:225–32. https://doi.org/10.1002/jcp.1041180302 [PubMed]
- 26. Timchenko NA, Wilde M, Kosai KI, Heydari A, Bilyeu TA, Finegold MJ, Mohamedali K, Richardson A, Darlington GJ. Regenerating livers of old rats contain high levels of C/EBPalpha that correlate with altered expression of cell cycle associated proteins. Nucleic Acids Res. 1998; 26:3293–99. https://doi.org/10.1093/nar/26.13.3293 [PubMed]
- 27. Iakova P, Awad SS, Timchenko NA. Aging reduces proliferative capacities of liver by switching pathways of C/EBPalpha growth arrest. Cell. 2003; 113:495–506. https://doi.org/10.1016/S0092-8674(03)00318-0 [PubMed]
- 28. Fortner JG, Lincer RM. Hepatic resection in the elderly. Ann Surg. 1990; 211:141–45. https://doi.org/10.1097/00000658-199002000-00005 [PubMed]
- 29. Timchenko NA. Aging and liver regeneration. Trends Endocrinol Metab. 2009; 20:171–76. https://doi.org/10.1016/j.tem.2009.01.005 [PubMed]
- 30. Wang X, Quail E, Hung NJ, Tan Y, Ye H, Costa RH. Increased levels of forkhead box M1B transcription factor in transgenic mouse hepatocytes prevent age-related proliferation defects in regenerating liver. Proc Natl Acad Sci USA. 2001; 98:11468–73. https://doi.org/10.1073/pnas.201360898 [PubMed]
- 31. Gagliano N, Grizzi F, Annoni G. Mechanisms of aging and liver functions. Dig Dis. 2007; 25:118–23. https://doi.org/10.1159/000099475 [PubMed]
- 32. Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG, Roebuck KA, Costa RH. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol. 1997; 17:1626–41. https://doi.org/10.1128/MCB.17.3.1626 [PubMed]
- 33. Ye H, Holterman AX, Yoo KW, Franks RR, Costa RH. Premature expression of the winged helix transcription factor HFH-11B in regenerating mouse liver accelerates hepatocyte entry into S phase. Mol Cell Biol. 1999; 19:8570–80. https://doi.org/10.1128/MCB.19.12.8570 [PubMed]
- 34. Ly DH, Lockhart DJ, Lerner RA, Schultz PG. Mitotic misregulation and human aging. Science. 2000; 287:2486–92. https://doi.org/10.1126/science.287.5462.2486 [PubMed]
- 35. Wang X, Hung NJ, Costa RH. Earlier expression of the transcription factor HFH-11B diminishes induction of p21(CIP1/WAF1) levels and accelerates mouse hepatocyte entry into S-phase following carbon tetrachloride liver injury. Hepatology. 2001; 33:1404–14. https://doi.org/10.1053/jhep.2001.24666 [PubMed]
- 36. Birkenmeier EH, Gwynn B, Howard S, Jerry J, Gordon JI, Landschulz WH, McKnight SL. Tissue-specific expression, developmental regulation, and genetic mapping of the gene encoding CCAAT/enhancer binding protein. Genes Dev. 1989; 3:1146–56. https://doi.org/10.1101/gad.3.8.1146 [PubMed]
- 37. Timchenko NA, Harris TE, Wilde M, Bilyeu TA, Burgess-Beusse BL, Finegold MJ, Darlington GJ. CCAAT/enhancer binding protein alpha regulates p21 protein and hepatocyte proliferation in newborn mice. Mol Cell Biol. 1997; 17:7353–61. https://doi.org/10.1128/MCB.17.12.7353 [PubMed]
- 38. Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ, Timchenko NA. C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell. 2001; 8:817–28. https://doi.org/10.1016/S1097-2765(01)00366-5 [PubMed]
- 39. Wang H, Goode T, Iakova P, Albrecht JH, Timchenko NA. C/EBPalpha triggers proteasome-dependent degradation of cdk4 during growth arrest. EMBO J. 2002; 21:930–41. https://doi.org/10.1093/emboj/21.5.930 [PubMed]
- 40. Arora V, Knapp DC, Smith BL, Statdfield ML, Stein DA, Reddy MT, Weller DD, Iversen PL. c-Myc antisense limits rat liver regeneration and indicates role for c-Myc in regulating cytochrome P-450 3A activity. J Pharmacol Exp Ther. 2000; 292:921–28. [PubMed]
- 41. Pedersen TA, Kowenz-Leutz E, Leutz A, Nerlov C. Cooperation between C/EBPalpha TBP/TFIIB and SWI/SNF recruiting domains is required for adipocyte differentiation. Genes Dev. 2001; 15:3208–16. https://doi.org/10.1101/gad.209901 [PubMed]
- 42. Tomiya T, Ogata I, Fujiwara K. Transforming growth factor α levels in liver and blood correlate better than hepatocyte growth factor with hepatocyte proliferation during liver regeneration. Am J Pathol. 1998; 153:955–61. https://doi.org/10.1016/S0002-9440(10)65637-4 [PubMed]
- 43. Shiota G, Wang TC, Nakamura T, Schmidt EV. Hepatocyte growth factor in transgenic mice: effects on hepatocyte growth, liver regeneration and gene expression. Hepatology. 1994; 19:962–72. https://doi.org/10.1002/hep.1840190423 [PubMed]
- 44. Enkhbold C, Morine Y, Utsunomiya T, Imura S, Ikemoto T, Arakawa Y, Saito Y, Yamada S, Ishikawa D, Shimada M. Dysfunction of liver regeneration in aged liver after partial hepatectomy. J Gastroenterol Hepatol. 2015; 30:1217–24. https://doi.org/10.1111/jgh.12930 [PubMed]
- 45. Pibiri M, Sulas P, Leoni VP, Perra A, Kowalik MA, Cordella A, Saggese P, Nassa G, Ravo M. Global gene expression profile of normal and regenerating liver in young and old mice. Age (Dordr). 2015; 37:9796. https://doi.org/10.1007/s11357-015-9796-7 [PubMed]
- 46. Ikawa-Yoshida A, Matsumoto T, Okano S, Aoyagi Y, Matsubara Y, Furuyama T, Nakatsu Y, Tsuzuki T, Onimaru M, Ohkusa T, Nomura M, Maehara Y. BubR1 Insufficiency Impairs Liver Regeneration in Aged Mice after Hepatectomy through Intercalated Disc Abnormality. Sci Rep. 2016; 6:32399. https://doi.org/10.1038/srep32399 [PubMed]
- 47. Yu H. Regulation of APC-Cdc20 by the spindle checkpoint. Curr Opin Cell Biol. 2002; 14:706–14. https://doi.org/10.1016/S0955-0674(02)00382-4 [PubMed]
- 48. Bolanos-Garcia VM, Blundell TL. BUB1 and BUBR1: multifaceted kinases of the cell cycle. Trends Biochem Sci. 2011; 36:141–50. https://doi.org/10.1016/j.tibs.2010.08.004 [PubMed]
- 49. Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004; 36:744–49. https://doi.org/10.1038/ng1382 [PubMed]
- 50. Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederländer NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, Eberhardt NL, Terzic A, van Deursen JM. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008; 10:825–36. https://doi.org/10.1038/ncb1744 [PubMed]
- 51. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–36. https://doi.org/10.1038/nature10600 [PubMed]
- 52. Matsumoto T, Baker DJ, d’Uscio LV, Mozammel G, Katusic ZS, van Deursen JM. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke. 2007; 38:1050–56. https://doi.org/10.1161/01.STR.0000257967.86132.01 [PubMed]
- 53. Kyuragi R, Matsumoto T, Harada Y, Saito S, Onimaru M, Nakatsu Y, Tsuzuki T, Nomura M, Yonemitsu Y, Maehara Y. BubR1 insufficiency inhibits neointimal hyperplasia through impaired vascular smooth muscle cell proliferation in mice. Arterioscler Thromb Vasc Biol. 2015; 35:341–47. https://doi.org/10.1161/ATVBAHA.114.304737 [PubMed]
- 54. Donetti E, Boschini E, Cerini A, Selleri S, Rumio C, Barajon I. Desmocollin 1 expression and desmosomal remodeling during terminal differentiation of human anagen hair follicle: an electron microscopic study. Exp Dermatol. 2004; 13:289–97. https://doi.org/10.1111/j.0906-6705.2004.00152.x [PubMed]
- 55. Bosch J, Pizcueta P, Feu F, Fernández M, García-Pagán JC. Pathophysiology of portal hypertension. Gastroenterol Clin North Am. 1992; 21:1–14. [PubMed]
- 56. Collins NL, Reginato MJ, Paulus JK, Sgroi DC, Labaer J, Brugge JS. G1/S cell cycle arrest provides anoikis resistance through Erk-mediated Bim suppression. Mol Cell Biol. 2005; 25:5282–91. https://doi.org/10.1128/MCB.25.12.5282-5291.2005 [PubMed]
- 57. Yang S, Zhang L, Chen X, Chen Y, Dong J. Oncoprotein YAP regulates the spindle checkpoint activation in a mitotic phosphorylation-dependent manner through up-regulation of BubR1. J Biol Chem. 2015; 290:6191–202. https://doi.org/10.1074/jbc.M114.624411 [PubMed]
- 58. Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 1994; 9:2145–52. [PubMed]
- 59. Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, Hisaminato A, Fujiwara T, Ito Y, Cantley LC, Yaffe MB. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000; 19:6778–91. https://doi.org/10.1093/emboj/19.24.6778 [PubMed]
- 60. Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010; 24:862–74. https://doi.org/10.1101/gad.1909210 [PubMed]
- 61. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010; 19:491–505. https://doi.org/10.1016/j.devcel.2010.09.011 [PubMed]
- 62. Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015; 163:811–28. https://doi.org/10.1016/j.cell.2015.10.044 [PubMed]
- 63. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007; 17:2054–60. https://doi.org/10.1016/j.cub.2007.10.039 [PubMed]
- 64. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007; 130:1120–33. https://doi.org/10.1016/j.cell.2007.07.019 [PubMed]
- 65. Grijalva JL, Huizenga M, Mueller K, Rodriguez S, Brazzo J, Camargo F, Sadri-Vakili G, Vakili K. Dynamic alterations in Hippo signaling pathway and YAP activation during liver regeneration. Am J Physiol Gastrointest Liver Physiol. 2014; 307:G196–204. https://doi.org/10.1152/ajpgi.00077.2014 [PubMed]
- 66. Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, Dong J. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. 2013; 73:6722–33. https://doi.org/10.1158/0008-5472.CAN-13-2049 [PubMed]
- 67. Gaspar P, Tapon N. Sensing the local environment: actin architecture and Hippo signalling. Curr Opin Cell Biol. 2014; 31:74–83. https://doi.org/10.1016/j.ceb.2014.09.003 [PubMed]
- 68. Loforese G, Malinka T, Keogh A, Baier F, Simillion C, Montani M, Halazonetis TD, Candinas D, Stroka D. Impaired liver regeneration in aged mice can be rescued by silencing Hippo core kinases MST1 and MST2. EMBO Mol Med. 2017; 9:46–60. https://doi.org/10.15252/emmm.201506089 [PubMed]
- 69. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007; 21:2747–61. https://doi.org/10.1101/gad.1602907 [PubMed]
- 70. Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011; 138:3907–14. https://doi.org/10.1242/dev.070987 [PubMed]
- 71. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011; 474:179–83. https://doi.org/10.1038/nature10137 [PubMed]
- 72. Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013; 154:1047–59. https://doi.org/10.1016/j.cell.2013.07.042 [PubMed]
- 73. Finch-Edmondson M, Sudol M. Framework to function: mechanosensitive regulators of gene transcription. Cell Mol Biol Lett. 2016; 21:28. https://doi.org/10.1186/s11658-016-0028-7 [PubMed]
- 74. Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M, Sheetz M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 2014; 588:2663–70. https://doi.org/10.1016/j.febslet.2014.04.012 [PubMed]
- 75. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, Fu XD, Mills GB, Guan KL. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012; 150:780–91. https://doi.org/10.1016/j.cell.2012.06.037 [PubMed]
- 76. Miller E, Yang J, DeRan M, Wu C, Su AI, Bonamy GM, Liu J, Peters EC, Wu X. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem Biol. 2012; 19:955–62. https://doi.org/10.1016/j.chembiol.2012.07.005 [PubMed]
- 77. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012; 26:54–68. https://doi.org/10.1101/gad.173435.111 [PubMed]
- 78. Ardestani A, Paroni F, Azizi Z, Kaur S, Khobragade V, Yuan T, Frogne T, Tao W, Oberholzer J, Pattou F, Conte JK, Maedler K. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med. 2014; 20:385–97. https://doi.org/10.1038/nm.3482 [PubMed]
- 79. Del Re DP, Matsuda T, Zhai P, Maejima Y, Jain MR, Liu T, Li H, Hsu CP, Sadoshima J. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol Cell. 2014; 54:639–50. https://doi.org/10.1016/j.molcel.2014.04.007 [PubMed]
- 80. Chao Y, Wang Y, Liu X, Ma P, Shi Y, Gao J, Shi Q, Hu J, Yu R, Zhou X. Mst1 regulates glioma cell proliferation via the AKT/mTOR signaling pathway. J Neurooncol. 2015; 121:279–88. https://doi.org/10.1007/s11060-014-1654-4 [PubMed]
- 81. Ledda-Columbano GM, Pibiri M, Cossu C, Molotzu F, Locker J, Columbano A. Aging does not reduce the hepatocyte proliferative response of mice to the primary mitogen TCPOBOP. Hepatology. 2004; 40:981–88. https://doi.org/10.1002/hep.1840400429 [PubMed]
- 82. Bellet MM, Sassone-Corsi P. Mammalian circadian clock and metabolism - the epigenetic link. J Cell Sci. 2010; 123:3837–48. https://doi.org/10.1242/jcs.051649 [PubMed]
- 83. Hastings MH, Reddy AB, Maywood ES. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci. 2003; 4:649–61. https://doi.org/10.1038/nrn1177 [PubMed]
- 84. Hardin PE, Hall JC, Rosbash M. Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature. 1990; 343:536–40. https://doi.org/10.1038/343536a0 [PubMed]
- 85. Lowrey PL, Takahashi JS. Genetics of the mammalian circadian system: photic entrainment, circadian pacemaker mechanisms, and posttranslational regulation. Annu Rev Genet. 2000; 34:533–62. https://doi.org/10.1146/annurev.genet.34.1.533 [PubMed]
- 86. Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002; 418:935–41. https://doi.org/10.1038/nature00965 [PubMed]
- 87. Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002; 109:307–20. https://doi.org/10.1016/S0092-8674(02)00722-5 [PubMed]
- 88. Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002; 417:78–83. https://doi.org/10.1038/nature744 [PubMed]
- 89. Kondratova AA, Kondratov RV. The circadian clock and pathology of the ageing brain. Nat Rev Neurosci. 2012; 13:325–35. https://doi.org/10.1038/nrn3208 [PubMed]
- 90. Nakamura TJ, Nakamura W, Yamazaki S, Kudo T, Cutler T, Colwell CS, Block GD. Age-related decline in circadian output. J Neurosci. 2011; 31:10201–05. https://doi.org/10.1523/JNEUROSCI.0451-11.2011 [PubMed]
- 91. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006; 20:1868–73. https://doi.org/10.1101/gad.1432206 [PubMed]
- 92. Cartee GD, Hepple RT, Bamman MM, Zierath JR. Exercise Promotes Healthy Aging of Skeletal Muscle. Cell Metab. 2016; 23:1034–47. https://doi.org/10.1016/j.cmet.2016.05.007 [PubMed]
- 93. Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell. 2015; 161:106–18. https://doi.org/10.1016/j.cell.2015.02.020 [PubMed]
- 94. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012; 13:225–38. https://doi.org/10.1038/nrm3293 [PubMed]
- 95. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014; 24:464–71. https://doi.org/10.1016/j.tcb.2014.04.002 [PubMed]
- 96. Ocampo A, Reddy P, Belmonte JC. Anti-Aging Strategies Based on Cellular Reprogramming. Trends Mol Med. 2016; 22:725–38. https://doi.org/10.1016/j.molmed.2016.06.005 [PubMed]
- 97. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015; 16:258–64. https://doi.org/10.1038/nrm3931 [PubMed]
- 98. Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, Dubeau L, Yap LP, Park R, Vinciguerra M, Di Biase S, Mirzaei H, Mirisola MG, et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015; 22:86–99. https://doi.org/10.1016/j.cmet.2015.05.012 [PubMed]
- 99. Grandison RC, Piper MD, Partridge L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature. 2009; 462:1061–64. https://doi.org/10.1038/nature08619 [PubMed]
- 100. Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999; 285:1390–93. https://doi.org/10.1126/science.285.5432.1390 [PubMed]
- 101. Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002; 418:344–48. https://doi.org/10.1038/nature00829 [PubMed]
- 102. Longo VD, Panda S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016; 23:1048–59. https://doi.org/10.1016/j.cmet.2016.06.001 [PubMed]
- 103. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009; 460:587–91. https://doi.org/10.1038/nature08197 [PubMed]
- 104. Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family’s role in aging and age-associated pathologies. J Clin Invest. 2013; 123:973–79. https://doi.org/10.1172/JCI64094 [PubMed]
- 105. Choi JE, Mostoslavsky R. Sirtuins, metabolism, and DNA repair. Curr Opin Genet Dev. 2014; 26:24–32. https://doi.org/10.1016/j.gde.2014.05.005 [PubMed]
- 106. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997; 389:349–52. https://doi.org/10.1038/38664 [PubMed]
- 107. Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998; 12:599–606. https://doi.org/10.1101/gad.12.5.599 [PubMed]
- 108. Wade PA, Wolffe AP. Histone acetyltransferases in control. Curr Biol. 1997; 7:R82–84. https://doi.org/10.1016/S0960-9822(06)00042-X [PubMed]
- 109. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001; 107:137–48. https://doi.org/10.1016/S0092-8674(01)00524-4 [PubMed]
- 110. Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005; 18:601–07. https://doi.org/10.1016/j.molcel.2005.04.021 [PubMed]
- 111. Martínez-Balbás MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. EMBO J. 2000; 19:662–71. https://doi.org/10.1093/emboj/19.4.662 [PubMed]
- 112. Feldman JL, Dittenhafer-Reed KE, Kudo N, Thelen JN, Ito A, Yoshida M, Denu JM. Kinetic and Structural Basis for Acyl-Group Selectivity and NAD(+) Dependence in Sirtuin-Catalyzed Deacylation. Biochemistry. 2015; 54:3037–50. https://doi.org/10.1021/acs.biochem.5b00150 [PubMed]
- 113. Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006; 75:435–65. https://doi.org/10.1146/annurev.biochem.74.082803.133500 [PubMed]
- 114. Verdin E. The many faces of sirtuins: coupling of NAD metabolism, sirtuins and lifespan. Nat Med. 2014; 20:25–27. https://doi.org/10.1038/nm.3447 [PubMed]
- 115. Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2014; 25:138–45. https://doi.org/10.1016/j.tem.2013.12.001 [PubMed]
- 116. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010; 5:253–95. https://doi.org/10.1146/annurev.pathol.4.110807.092250 [PubMed]
- 117. Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A, Easlon EJ, Lin SJ, Guarente L. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006; 4:e31. https://doi.org/10.1371/journal.pbio.0040031 [PubMed]
- 118. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004; 303:2011–15. https://doi.org/10.1126/science.1094637 [PubMed]
- 119. Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004; 116:551–63. https://doi.org/10.1016/S0092-8674(04)00126-6 [PubMed]
- 120. Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007; 28:91–106. https://doi.org/10.1016/j.molcel.2007.07.032 [PubMed]
- 121. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005; 434:113–18. https://doi.org/10.1038/nature03354 [PubMed]
- 122. Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005; 310:1641. https://doi.org/10.1126/science.1118357 [PubMed]
- 123. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004; 305:390–92. https://doi.org/10.1126/science.1099196 [PubMed]
- 124. Orozco-Solis R, Ramadori G, Coppari R, Sassone-Corsi P. SIRT1 Relays Nutritional Inputs to the Circadian Clock Through the Sf1 Neurons of the Ventromedial Hypothalamus. Endocrinology. 2015; 156:2174–84. https://doi.org/10.1210/en.2014-1805 [PubMed]
- 125. Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006; 49:191–203. https://doi.org/10.1016/j.neuron.2005.12.021 [PubMed]
- 126. Ramadori G, Fujikawa T, Anderson J, Berglund ED, Frazao R, Michán S, Vianna CR, Sinclair DA, Elias CF, Coppari R. SIRT1 deacetylase in SF1 neurons protects against metabolic imbalance. Cell Metab. 2011; 14:301–12. https://doi.org/10.1016/j.cmet.2011.06.014 [PubMed]
- 127. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014; 24:464–71. https://doi.org/10.1016/j.tcb.2014.04.002 [PubMed]
- 128. Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013; 153:1448–60. https://doi.org/10.1016/j.cell.2013.05.027 [PubMed]
- 129. Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008; 134:329–40. https://doi.org/10.1016/j.cell.2008.07.002 [PubMed]
- 130. Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013; 27:2072–85. https://doi.org/10.1101/gad.227439.113 [PubMed]
- 131. Sato S, Solanas G, Peixoto FO, Bee L, Symeonidi A, Schmidt MS, Brenner C, Masri S, Benitah SA, Sassone-Corsi P. Circadian reprogramming in the liver identifies metabolic pathways of aging. Cell. 2017; 170:664–677.e11. https://doi.org/10.1016/j.cell.2017.07.042 [PubMed]
- 132. Solanas G, Oliveira Peixoto F, Perdiguero E, Jardı´ M, Ruiz-Bonilla V, Datta D, Symeonidi A, Castellanos A, Welz P, Martìn Caballero J, Sassone-Corsi P, Muñoz-Cánoves P, Benitah SA. Adult stem cells undergo circadian reprogramming during ageing. Cell. 2017; 170:678–92. https://doi.org/10.1016/j.cell.2017.07.035 [PubMed]
- 133. Hagopian K, Ramsey JJ, Weindruch R. Caloric restriction increases gluconeogenic and transaminase enzyme activities in mouse liver. Exp Gerontol. 2003; 38:267–78. https://doi.org/10.1016/S0531-5565(02)00202-4 [PubMed]
- 134. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015; 21:805–21. https://doi.org/10.1016/j.cmet.2015.05.014 [PubMed]
- 135. Seufert CD, Graf M, Janson G, Kuhn A, Söling HD. Formation of free acetate by isolated perfused livers from normal, starved and diabetic rats. Biochem Biophys Res Commun. 1974; 57:901–09. https://doi.org/10.1016/0006-291X(74)90631-7 [PubMed]
- 136. Masri S, Sassone-Corsi P. Sirtuins and the circadian clock: bridging chromatin and metabolism. Sci Signal. 2014; 7:re6. https://doi.org/10.1126/scisignal.2005685 [PubMed]
- 137. Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA. 2006; 103:10230–35. https://doi.org/10.1073/pnas.0604392103 [PubMed]
- 138. Sahar S, Masubuchi S, Eckel-Mahan K, Vollmer S, Galla L, Ceglia N, Masri S, Barth TK, Grimaldi B, Oluyemi O, Astarita G, Hallows WC, Piomelli D, et al. Circadian control of fatty acid elongation by SIRT1 protein-mediated deacetylation of acetyl-coenzyme A synthetase 1. J Biol Chem. 2014; 289:6091–97. https://doi.org/10.1074/jbc.M113.537191 [PubMed]
- 139. Sahar S, Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nat Rev Cancer. 2009; 9:886–96. https://doi.org/10.1038/nrc2747 [PubMed]
- 140. Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002; 111:41–50. https://doi.org/10.1016/S0092-8674(02)00961-3 [PubMed]
- 141. Gery S, Komatsu N, Baldjyan L, Yu A, Koo D, Koeffler HP. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol Cell. 2006; 22:375–82. https://doi.org/10.1016/j.molcel.2006.03.038 [PubMed]
- 142. Kang TH, Reardon JT, Kemp M, Sancar A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci USA. 2009; 106:2864–67. https://doi.org/10.1073/pnas.0812638106 [PubMed]
- 143. Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003; 302:255–59. https://doi.org/10.1126/science.1086271 [PubMed]
- 144. Bellet MM, Masri S, Astarita G, Sassone-Corsi P, Della Fazia MA, Servillo G. Histone Deacetylase SIRT1 Controls Proliferation, Circadian Rhythm, and Lipid Metabolism during Liver Regeneration in Mice. J Biol Chem. 2016; 291:23318–29. https://doi.org/10.1074/jbc.M116.737114 [PubMed]
- 145. Singh CK, George J, Nihal M, Sabat G, Kumar R, Ahmad N. Novel downstream molecular targets of SIRT1 in melanoma: a quantitative proteomics approach. Oncotarget. 2014; 5:1987–99. https://doi.org/10.18632/oncotarget.1898 [PubMed]
- 146. Pibiri M, Ledda-Columbano GM, Cossu C, Simbula G, Menegazzi M, Shinozuka H, Columbano A. Cyclin D1 is an early target in hepatocyte proliferation induced by thyroid hormone (T3). FASEB J. 2001; 15:1006–13. https://doi.org/10.1096/fj.00-0416com [PubMed]
- 147. Ledda-Columbano GM, Pibiri M, Loi R, Perra A, Shinozuka H, Columbano A. Early increase in cyclin D1 expression and accelerated entry of mouse hepatocytes into S phase after administration of the mitogen 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene.A m. J Pathol. 2000; 56:91–97.
- 148. Ledda-Columbano GM, Curto M, Piga R, Zedda AI, Menegazzi M, Sartori C, Shinozuka H, Bluethmann H, Poli V, Ciliberto G, Columbano A. In vivo hepatocyte proliferation is inducible through a TNF and IL-6-independent pathway. Oncogene. 1998; 17:1039–44. https://doi.org/10.1038/sj.onc.1202018 [PubMed]
- 149. Goldsworthy TL, Goldsworthy SM, Sprankle CS, Butterworth BE. Expression of myc, fos and Ha-ras associated with chemically induced cell proliferation in the rat liver. Cell Prolif. 1994; 27:269–78. https://doi.org/10.1111/j.1365-2184.1994.tb01424.x [PubMed]
- 150. Skrtic S, Ekberg S, Wallenius V, Enerbäck S, Hedin L, Jansson JO. Changes in expression of CCAAT/enhancer binding protein alpha (C/EBP alpha) and C/EBP beta in rat liver after partial hepatectomy but not after treatment with cyproterone acetate. J Hepatol. 1997; 27:903–11. https://doi.org/10.1016/S0168-8278(97)80329-7 [PubMed]
- 151. Leoni VP, Ledda-Columbano GM, Pibiri M, Saliba C, Perra A, Kowalik MA, Grober OM, Ravo M, Weisz A, Locker J, Ghiso E, Giordano S, Columbano A. Expression of c-jun is not mandatory for mouse hepatocyte proliferation induced by two nuclear receptor ligands: TCPOBOP and T3. J Hepatol. 2011; 55:1069–78. https://doi.org/10.1016/j.jhep.2011.02.016 [PubMed]
- 152. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005; 433:760–64. https://doi.org/10.1038/nature03260 [PubMed]
- 153. Krupczak-Hollis K, Wang X, Dennewitz MB, Costa RH. Growth hormone stimulates proliferation of old-aged regenerating liver through forkhead box m1b. Hepatology. 2003; 38:1552–62. https://doi.org/10.1053/jhep.2003.08052 [PubMed]
- 154. Purdom S, Chen QM. p66(Shc): at the crossroad of oxidative stress and the genetics of aging. Trends Mol Med. 2003; 9:206–10. https://doi.org/10.1016/S1471-4914(03)00048-0 [PubMed]
- 155. Singh P, Goode T, Dean A, Awad SS, Darlington GJ. Elevated interferon gamma signaling contributes to impaired regeneration in the aged liver. J Gerontol A Biol Sci Med Sci. 2011; 66:944–56. https://doi.org/10.1093/gerona/glr094 [PubMed]
- 156. Moolten FL, Oakman NJ, Bucher NL. Accelerated response of hepatic DNA synthesis to partial hepatectomy in rats pretreated with growth hormone or surgical stress. Cancer Res. 1970; 30:2353–57. [PubMed]
- 157. Jin J, Wang GL, Shi X, Darlington GJ, Timchenko NA. The age-associated decline of glycogen synthase kinase 3beta plays a critical role in the inhibition of liver regeneration. Mol Cell Biol. 2009; 29:3867–80. https://doi.org/10.1128/MCB.00456-09 [PubMed]
- 158. Wang GL, Shi X, Salisbury E, Sun Y, Albrecht JH, Smith RG, Timchenko NA. Cyclin D3 maintains growth-inhibitory activity of C/EBPalpha by stabilizing C/EBPalpha-cdk2 and C/EBPalpha-Brm complexes. Mol Cell Biol. 2006; 26:2570–82. https://doi.org/10.1128/MCB.26.7.2570-2582.2006 [PubMed]
- 159. Pellegrini M, Pacini S, Baldari CT. p66SHC: the apoptotic side of Shc proteins. Apoptosis. 2005; 10:13–18. https://doi.org/10.1007/s10495-005-6057-8 [PubMed]
- 160. Bonfini L, Migliaccio E, Pelicci G, Lanfrancone L, Pelicci PG. Not all Shc’s roads lead to Ras. Trends Biochem Sci. 1996; 21:257–61. https://doi.org/10.1016/S0968-0004(96)10033-5 [PubMed]
- 161. Purdom S, Chen QM. Linking oxidative stress and genetics of aging with p66Shc signaling and forkhead transcription factors. Biogerontology. 2003; 4:181–91. https://doi.org/10.1023/A:1025123413403 [PubMed]
- 162. Ravichandran KS. Signaling via Shc family adapter proteins. Oncogene. 2001; 20:6322–30. https://doi.org/10.1038/sj.onc.1204776 [PubMed]
- 163. Haga S, Terui K, Fukai M, Oikawa Y, Irani K, Furukawa H, Todo S, Ozaki M. Preventing hypoxia/reoxygenation damage to hepatocytes by p66(shc) ablation: up-regulation of anti-oxidant and anti-apoptotic proteins. J Hepatol. 2008; 48:422–32. https://doi.org/10.1016/j.jhep.2007.11.018 [PubMed]
- 164. Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002; 295:2450–52. https://doi.org/10.1126/science.1069004 [PubMed]
- 165. Zaccagnini G, Martelli F, Fasanaro P, Magenta A, Gaetano C, Di Carlo A, Biglioli P, Giorgio M, Martin-Padura I, Pelicci PG, Capogrossi MC. p66ShcA modulates tissue response to hindlimb ischemia. Circulation. 2004; 109:2917–23. https://doi.org/10.1161/01.CIR.0000129309.58874.0F [PubMed]
- 166. Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999; 402:309–13. https://doi.org/10.1038/46311 [PubMed]
- 167. Haga S, Morita N, Irani K, Fujiyoshi M, Ogino T, Ozawa T, Ozaki M. p66(Shc) has a pivotal function in impaired liver regeneration in aged mice by a redox-dependent mechanism. Lab Invest. 2010; 90:1718–26. https://doi.org/10.1038/labinvest.2010.119 [PubMed]
- 168. Brüünsgaard H, Pedersen BK. Age-related inflammatory cytokines and disease. Immunol Allergy Clin North Am. 2003; 23:15–39. https://doi.org/10.1016/S0889-8561(02)00056-5 [PubMed]
- 169. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000; 908:244–54. https://doi.org/10.1111/j.1749-6632.2000.tb06651.x [PubMed]
- 170. Singh P, Coskun ZZ, Goode C, Dean A, Thompson-Snipes L, Darlington G. Lymphoid neogenesis and immune infiltration in aged liver. Hepatology. 2008; 47:1680–90. https://doi.org/10.1002/hep.22224 [PubMed]
- 171. Tura BJ, Bunyan KE, Harrison DJ. The effect of IFNgamma on the hepatocyte: cell cycle and apoptosis. Int J Exp Pathol. 2001; 82:317–26. https://doi.org/10.1046/j.1365-2613.2001.00207.x [PubMed]
- 172. Kano A, Haruyama T, Akaike T, Watanabe Y. IRF-1 is an essential mediator in IFN-gamma-induced cell cycle arrest and apoptosis of primary cultured hepatocytes. Biochem Biophys Res Commun. 1999; 257:672–77. https://doi.org/10.1006/bbrc.1999.0276 [PubMed]
- 173. Kano A, Watanabe Y, Takeda N, Aizawa S, Akaike T. Analysis of IFN-gamma-induced cell cycle arrest and cell death in hepatocytes. J Biochem. 1997; 121:677–83. https://doi.org/10.1093/oxfordjournals.jbchem.a021639 [PubMed]
- 174. Brooling JT, Campbell JS, Mitchell C, Yeoh GC, Fausto N. Differential regulation of rodent hepatocyte and oval cell proliferation by interferon gamma. Hepatology. 2005; 41:906–15. https://doi.org/10.1002/hep.20645 [PubMed]
- 175. Lee HJ, Oh YK, Rhee M, Lim JY, Hwang JY, Park YS, Kwon Y, Choi KH, Jo I, Park SI, Gao B, Kim WH. The role of STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial transmembrane potential during hepatic cell death induced by LPS/d-GalN. J Mol Biol. 2007; 369:967–84. https://doi.org/10.1016/j.jmb.2007.03.072 [PubMed]
- 176. Hofseth LJ, Saito S, Hussain SP, Espey MG, Miranda KM, Araki Y, Jhappan C, Higashimoto Y, He P, Linke SP, Quezado MM, Zurer I, Rotter V, et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci USA. 2003; 100:143–48. https://doi.org/10.1073/pnas.0237083100 [PubMed]
- 177. Singh P, Goode T, Dean A, Awad SS, Darlington GJ. Elevated interferon gamma signaling contributes to impaired regeneration in the aged liver. J Gerontol A Biol Sci Med Sci. 2011; 66:944–56. https://doi.org/10.1093/gerona/glr094 [PubMed]
- 178. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956; 11:298–300. https://doi.org/10.1093/geronj/11.3.298 [PubMed]
- 179. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000; 408:239–47. https://doi.org/10.1038/35041687 [PubMed]
- 180. Cheng Y, Wang X, Wang B, Zhou H, Dang S, Shi Y, Hao L, Luo Q, Jin M, Zhou Q, Zhang Y. Aging-associated oxidative stress inhibits liver progenitor cell activation in mice. Aging (Albany NY). 2017; 9:1359–74. https://doi.org/10.18632/aging.101232 [PubMed]
- 181. Farber E. Similarities in the sequence of early histological changes induced in the liver of the rat by ethionine, 2-acetylamino-fluorene, and 3′-methyl-4-dimethylaminoazobenzene. Cancer Res. 1956; 16:142–48. [PubMed]
- 182. Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004; 39:1477–87. https://doi.org/10.1002/hep.20214 [PubMed]
- 183. Español-Suñer R, Carpentier R, Van Hul N, Legry V, Achouri Y, Cordi S, Jacquemin P, Lemaigre F, Leclercq IA. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology. 2012; 143:1564–1575.e7. https://doi.org/10.1053/j.gastro.2012.08.024 [PubMed]
- 184. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, Sato T, Hamer K, Sasaki N, Finegold MJ, Haft A, Vries RG, Grompe M, Clevers H. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013; 494:247–50. https://doi.org/10.1038/nature11826 [PubMed]
- 185. Tanimizu N, Nishikawa Y, Ichinohe N, Akiyama H, Mitaka T. Sry HMG box protein 9-positive (Sox9+) epithelial cell adhesion molecule-negative (EpCAM-) biphenotypic cells derived from hepatocytes are involved in mouse liver regeneration. J Biol Chem. 2014; 289:7589–98. https://doi.org/10.1074/jbc.M113.517243 [PubMed]
- 186. Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, Thung SN, Wells RG, Greenbaum LE, Stanger BZ. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013; 27:719–24. https://doi.org/10.1101/gad.207803.112 [PubMed]
- 187. Feng D, Kong X, Weng H, Park O, Wang H, Dooley S, Gershwin ME, Gao B. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology. 2012; 143:188–98.e7. https://doi.org/10.1053/j.gastro.2012.03.044 [PubMed]
- 188. Jelnes P, Santoni-Rugiu E, Rasmussen M, Friis SL, Nielsen JH, Tygstrup N, Bisgaard HC. Remarkable heterogeneity displayed by oval cells in rat and mouse models of stem cell-mediated liver regeneration. Hepatology. 2007; 45:1462–70. https://doi.org/10.1002/hep.21569 [PubMed]
- 189. Wang Y, Huang X, He L, Pu W, Li Y, Liu Q, Li Y, Zhang L, Yu W, Zhao H, Zhou Y, Zhou B. Genetic tracing of hepatocytes in liver homeostasis, injury, and regeneration. J Biol Chem. 2017; 292:8594–604. https://doi.org/10.1074/jbc.M117.782029 [PubMed]
- 190. Rodrigo-Torres D, Affò S, Coll M, Morales-Ibanez O, Millán C, Blaya D, Alvarez-Guaita A, Rentero C, Lozano JJ, Maestro MA, Solar M, Arroyo V, Caballería J, et al. The biliary epithelium gives rise to liver progenitor cells. Hepatology. 2014; 60:1367–77. https://doi.org/10.1002/hep.27078 [PubMed]
- 191. Schaub JR, Malato Y, Gormond C, Willenbring H. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Reports. 2014; 8:933–39. https://doi.org/10.1016/j.celrep.2014.07.003 [PubMed]
- 192. Tarlow BD, Finegold MJ, Grompe M. Clonal tracing of Sox9+ liver progenitors in mouse oval cell injury. Hepatology. 2014; 60:278–89. https://doi.org/10.1002/hep.27084 [PubMed]
- 193. Yanger K, Knigin D, Zong Y, Maggs L, Gu G, Akiyama H, Pikarsky E, Stanger BZ. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell. 2014; 15:340–49. https://doi.org/10.1016/j.stem.2014.06.003 [PubMed]
- 194. Overturf K, al-Dhalimy M, Ou CN, Finegold M, Grompe M. Serial transplantation reveals the stem-cell-like regenerative potential of adult mouse hepatocytes. Am J Pathol. 1997; 151:1273–80. [PubMed]
- 195. Sekiya S, Suzuki A. Hepatocytes, rather than cholangiocytes, can be the major source of primitive ductules in the chronically injured mouse liver. Am J Pathol. 2014; 184:1468–78. https://doi.org/10.1016/j.ajpath.2014.01.005 [PubMed]
- 196. Marcos R, Correia-Gomes C. Long live the liver: immunohistochemical and stereological study of hepatocytes, liver sinusoidal endothelial cells, Kupffer cells and hepatic stellate cells of male and female rats throughout ageing. Cell Tissue Res. 2016; 366:639–49. https://doi.org/10.1007/s00441-016-2490-y [PubMed]
- 197. Schmucker DL, Sanchez H. Liver regeneration and aging: a current perspective. Curr Gerontol Geriatr Res. 2011; 2011:526379. https://doi.org/10.1155/2011/526379 [PubMed]
- 198. Warren A, Cogger VC, Fraser R, Deleve LD, McCuskey RS, Le Couteur DG. The effects of old age on hepatic stellate cells. Curr Gerontol Geriatr Res. 2011; 2011:439835. https://doi.org/10.1155/2011/439835 [PubMed]
- 199. Melton AC, Yee HF. Hepatic stellate cell protrusions couple platelet-derived growth factor-BB to chemotaxis. Hepatology. 2007; 45:1446–53. https://doi.org/10.1002/hep.21606 [PubMed]
- 200. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008; 88:125–72. https://doi.org/10.1152/physrev.00013.2007 [PubMed]
- 201. Saito Y, Morine Y, Shimada M. Mechanism of impairment on liver regeneration in elderly patients: role of hepatic stellate cell function. Hepatol Res. 2017; 47:505–13. https://doi.org/10.1111/hepr.12872 [PubMed]
- 202. Vasto S, Candore G, Balistreri CR, Caruso M, Colonna-Romano G, Grimaldi MP, Listi F, Nuzzo D, Lio D, Caruso C. Inflammatory networks in ageing, age-related diseases and longevity. Mech Ageing Dev. 2007; 128:83–91. https://doi.org/10.1016/j.mad.2006.11.015 [PubMed]
- 203. Ferrucci L, Ble A, Bandinelli S, Lauretani F, Suthers K, Guralnik JM. A flame burning within. Aging Clin Exp Res. 2004; 16:240–43. https://doi.org/10.1007/BF03327390 [PubMed]
- 204. Bruunsgaard H. The clinical impact of systemic low-level inflammation in elderly populations. With special reference to cardiovascular disease, dementia and mortality. Dan Med Bull. 2006; 53:285–309. [PubMed]
- 205. Maggio M, Guralnik JM, Longo DL, Ferrucci L. Interleukin-6 in aging and chronic disease: a magnificent pathway. J Gerontol A Biol Sci Med Sci. 2006; 61:575–84. https://doi.org/10.1093/gerona/61.6.575 [PubMed]
- 206. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008; 6:2853–68. https://doi.org/10.1371/journal.pbio.0060301 [PubMed]
- 207. Young AR, Narita M. SASP reflects senescence. EMBO Rep. 2009; 10:228–30. https://doi.org/10.1038/embor.2009.22 [PubMed]
- 208. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010; 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144 [PubMed]
- 209. Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005; 436:720–24. https://doi.org/10.1038/nature03890 [PubMed]
- 210. Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dörken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005; 436:660–65. https://doi.org/10.1038/nature03841 [PubMed]
- 211. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010; 10:51–57. https://doi.org/10.1038/nrc2772 [PubMed]
- 212. Wang MJ, Chen F, Li JX, Liu CC, Zhang HB, Xia Y, Yu B, You P, Xiang D, Lu L, Yao H, Borjigin U, Yang GS, et al. Reversal of hepatocyte senescence after continuous in vivo cell proliferation. Hepatology. 2014; 60:349–61. https://doi.org/10.1002/hep.27094 [PubMed]
- 213. Bhatia-Dey N, Kanherkar RR, Stair SE, Makarev EO, Csoka AB. Cellular Senescence as the Causal Nexus of Aging. Front Genet. 2016; 7:13. https://doi.org/10.3389/fgene.2016.00013 [PubMed]
- 214. Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dollé ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006; 441:1011–14. https://doi.org/10.1038/nature04844 [PubMed]
- 215. Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006; 311:1257. https://doi.org/10.1126/science.1122446 [PubMed]
- 216. Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006; 443:421–26. https://doi.org/10.1038/nature05159 [PubMed]
- 217. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006; 443:453–57. https://doi.org/10.1038/nature05092 [PubMed]
- 218. Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006; 443:448–52. https://doi.org/10.1038/nature05091 [PubMed]
- 219. Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007; 8:703–13. https://doi.org/10.1038/nrm2241 [PubMed]
- 220. Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E, González S, Serrano AL, Perdiguero E, Muñoz-Cánoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014; 506:316–21. https://doi.org/10.1038/nature13013 [PubMed]
- 221. García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Muñoz-Cánoves P. Autophagy maintains stemness by preventing senescence. Nature. 2016; 529:37–42. https://doi.org/10.1038/nature16187 [PubMed]
- 222. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016; 530:184–89. https://doi.org/10.1038/nature16932 [PubMed]
- 223. Hoare M, Das T, Alexander G. Ageing, telomeres, senescence, and liver injury. J Hepatol. 2010; 53:950–61. https://doi.org/10.1016/j.jhep.2010.06.009 [PubMed]
- 224. Zhu C, Ikemoto T, Utsunomiya T, Yamada S, Morine Y, Imura S, Arakawa Y, Takasu C, Ishikawa D, Shimada M. Senescence-related genes possibly responsible for poor liver regeneration after hepatectomy in elderly patients. J Gastroenterol Hepatol. 2014; 29:1102–08. https://doi.org/10.1111/jgh.12468 [PubMed]
- 225. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007; 8:729–40. https://doi.org/10.1038/nrm2233 [PubMed]
- 226. Vicente R, Mausset-Bonnefont AL, Jorgensen C, Louis-Plence P, Brondello JM. Cellular senescence impact on immune cell fate and function. Aging Cell. 2016; 15:400–06. https://doi.org/10.1111/acel.12455 [PubMed]
- 227. Kumar S, Millis AJ, Baglioni C. Expression of interleukin 1-inducible genes and production of interleukin 1 by aging human fibroblasts. Proc Natl Acad Sci USA. 1992; 89:4683–87. https://doi.org/10.1073/pnas.89.10.4683 [PubMed]
- 228. Wang S, Moerman EJ, Jones RA, Thweatt R, Goldstein S. Characterization of IGFBP-3, PAI-1 and SPARC mRNA expression in senescent fibroblasts. Mech Ageing Dev. 1996; 92:121–32. https://doi.org/10.1016/S0047-6374(96)01814-3 [PubMed]
- 229. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008; 133:1006–18. https://doi.org/10.1016/j.cell.2008.03.038 [PubMed]
- 230. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008; 134:657–67. https://doi.org/10.1016/j.cell.2008.06.049 [PubMed]
- 231. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M. Programmed cell senescence during mammalian embryonic development. Cell. 2013; 155:1104–18. https://doi.org/10.1016/j.cell.2013.10.019 [PubMed]
- 232. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, Keyes WM. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013; 155:1119–30. https://doi.org/10.1016/j.cell.2013.10.041 [PubMed]
- 233. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001; 98:12072–77. https://doi.org/10.1073/pnas.211053698 [PubMed]
- 234. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013; 499:97–101. https://doi.org/10.1038/nature12347 [PubMed]
- 235. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017; 31:172–83. https://doi.org/10.1101/gad.290635.116 [PubMed]
- 236. de Keizer PL. The Fountain of Youth by Targeting Senescent Cells? Trends Mol Med. 2017; 23:6–17. https://doi.org/10.1016/j.molmed.2016.11.006 [PubMed]
- 237. Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, Fernandez-Marcos PJ, Muñoz-Martin M, Blanco-Aparicio C, Pastor J, Gómez-López G, De Martino A, Blasco MA, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016; 354:6315. https://doi.org/10.1126/science.aaf4445 [PubMed]
- 238. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–36. https://doi.org/10.1038/nature10600 [PubMed]
- 239. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000; 290:1717–21. https://doi.org/10.1126/science.290.5497.1717 [PubMed]
- 240. Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010; 53:1123–34. https://doi.org/10.1016/j.jhep.2010.07.006 [PubMed]
- 241. Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010; 12:842–46. https://doi.org/10.1038/ncb0910-842 [PubMed]
- 242. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011; 146:682–95. https://doi.org/10.1016/j.cell.2011.07.030 [PubMed]
- 243. Schneider JL, Villarroya J, Diaz-Carretero A, Patel B, Urbanska AM, Thi MM, Villarroya F, Santambrogio L, Cuervo AM. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell. 2015; 14:249–64. https://doi.org/10.1111/acel.12310 [PubMed]
- 244. Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008; 14:959–65. https://doi.org/10.1038/nm.1851 [PubMed]
- 245. Liu A, Guo E, Yang J, Yang Y, Liu S, Jiang X, Hu Q, Dirsch O, Dahmen U, Zhang C, Gewirtz DA, Fang H. Young plasma reverses age-dependent alterations in hepatic function through the restoration of autophagy. Aging Cell. 2018; 17:e12708. https://doi.org/10.1111/acel.12708 [PubMed]
- 246. Columbano A, Ledda-Columbano GM. Mitogenesis by ligands of nuclear receptors: an attractive model for the study of the molecular mechanisms implicated in liver growth. Cell Death Differ. 2003 (Suppl 1); 10:S19–21. https://doi.org/10.1038/sj.cdd.4401113 [PubMed]