



Senolytics are drugs that extend lifespan and delay some age-related diseases by killing senescent cells [1–4]. In fact, drug screens have identified a diverse group of drugs that are preferentially toxic to at least some senescent cells in some cellular models [2–9]. So far, however, their selectivity against senescent cells is modest and cell-type-specific [8–11]. Nevertheless, targeting senescent cells has been shown in animal models to prevent such age-related pathologies as emphysema [12], lung fibrosis [13–15], atherosclerosis [16,17], osteoporosis [18], osteoarthritis [19,20], renal disease [21], intervertebral disk pathology [2], hepatic steatosis [22] and other age-related conditions [4,7,18,23,24].
In this editorial commentary, I want to draw your attention to the paradoxes associated with senolytics, which argue against the dogma that says aging is a functional decline caused by molecular damage. This dogma predicts that senolytics should accelerate aging. If aging is caused by loss of function, then killing senescent cells would be expected to accelerate aging, given that dead cells have no functionality at all. Instead, however, senolytics slow aging, which highlights a contradiction in the prevailing dogma.
The theory of hyperfunctional aging [25–32] addresses this paradox. Killing senescent cells is beneficial because senescent cells are hyperfunctional [33]. The hypersecretory phenotype or Senescence-Associated Secretory Phenotype (SASP) is the best-known example of universal hyperfunction [34–36]. Most such hyperfunctions are tissue-specific. For example, senescent beta cells overproduce insulin [37] and thus activate mTOR in hepatocytes, adipocytes and other cells, causing their hyperfunction, which in turn leads to metabolic syndrome (obesity, hypertension, hyperlipidemia and hyperglycemia) and is also a risk factor for cancer [38–40]. SASP, hyperinsulinemia and obesity, hypertension, hyperlipidemia and hyperglycemia are all examples of absolute hyperfunction (an increase in functionality). In comparison, relative hyperfunction is an insufficient decrease of unneeded function. For example, protein synthesis decreases with aging, but that decrease is not sufficient [30]. In analogy, a car moving on the highway at 65 mph is not “hyperfunctional.” But if the car were to exit the highway and enter a residential driveway at only 60 mph it would be “hyperfunctional,” and stopping that car would likely prevent damage to other objects. Similarly, killing hyperfunctional cells can prevent organismal damage. Senolytics eliminate hyperfunctional cells, which otherwise damage organs (Figure 1).
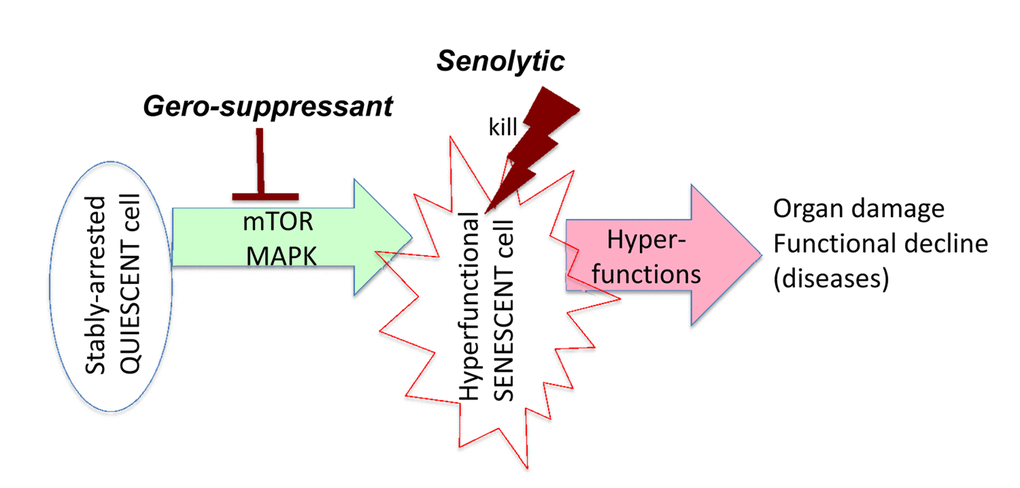
Figure 1. Target of senolytics in the aging quasi-program. In post-mitotic quiescent cells in an organism, growth-promoting effectors such as mTOR drive conversion to senescence. Hyperfunctional senescent cells activate other cells (including cells in distant organs), rendering them also hyperfunctional, which eventually leads to organ damage. This process manifests as functional decline, a terminal event secondary to initial hyperfunction. Senolytics such as ABT263 or 737 kill hyperfunctional senescent cells, preventing damage to organs. Gerosuppressants such as rapamycin suppress geroconversion and may decrease hyperfunction of already senescent cells, thereby slowing disease progression (not shown here in scheme).
Senolytics should not be confused with gerosuppressants (Figure 1). Gerosuppressants, such as rapamycin, do not kill cells; they instead prevent cellular conversion to senescence (geroconversion) [33]. Rapamycin also slows disease progression by limiting the hyperfunction of senescent cells. Notably, some senolytics are also gerosuppressants. For example, inhibitors of MEK [41–43] or PI3K [2,41] are both gerosuppressants [41] and senolytics [2,42,43].
It may seem paradoxical that senolytics are anticancer drugs [44] because standard anticancer agents cause molecular damage. According to the hyperfunction theory [45], molecular damage does not cause aging. Although accumulation of molecular damage does happen and would destroy the organism eventually, no organism lives long enough for that to occur because TOR-driven (hyperfunctional) aging kills it first. If TOR-driven aging (i.e., aging as we currently know it) were abolished, then organisms would die from “post-aging syndrome” due to molecular damage (see Figure 8 in ref. [25].). Molecular damage contributes to some age-related diseases. But these diseases would arise even without molecular damage [45]. Molecular damage is essential for most types of cancer, but a senescent microenvironment [46] and overall organism aging (and associated diseases such as diabetes) also play roles [47], as does clonal selection for mTOR activation in cancer cells [48]. Importantly, molecular damage renders cancer cells robust and hyperfunctional. Cancer cells kill an organism not because molecular damage makes them weak; it is because the molecular damage makes them robust and hyperfunctional. If accumulation of molecular damage leads to immortalization and robustness, then aging cannot represent functional decline caused by molecular damage [48].
All senolytics, without exception, were initially investigated or specifically developed as anticancer drugs. But not all anticancer drugs are senolytics. Both senolytics and gerosuppressants belong to a very special subgroup of oncotargeted drugs [49]. Various pathways involving IGF-1, Ras, MEK, AMPK, TSC1/2, FOXO, PI3K, mTOR, S6K, and NFκB comprise a mTOR-related network and are involved in aging [49]. Oncoproteins promote aging, while tumor suppressors are gerosuppressors, which inhibit aging [48,50]. As depicted a decade ago (see Figure 3 in ref. [51]. and Figures 4 and 9 in ref. [25].), oncotargets are gerotargets that are also mTOR activators, while tumor and aging suppressors are mTOR inhibitors. In brief, geroconversion and oncogenic transformation are two sides of the same process [50]. Gerogenic oncogenes activate the mTOR pathway, driving geroconversion of cell cycle-arrested cells. When cell cycle control is disabled, they drive oncogenic transformation [48,50].
Many puzzles remain. For example, killing senescent adipocytes, macrophages or foam cells will slow diseases such as atherosclerosis and metabolic diseases, and killing senescent glial cells can prevent cognitive decline [23]. On the other hand, killing some senescent cell types may be counterproductive. For example, killing senescent beta cells may lead to diabetes [37], and killing of senescent hyperfunctional neurons in Alzheimer’s disease may have unpredictable consequences. Fortunately, senolytics are tissue-specific and only kill some types of senescent cells [8–11], which may make them safer.
To add further complication to the paradoxes associated with senolytics, it was shown that many detected p16/β-gal-positive cells are not senescent cells, but are instead hyperfunctional macrophages, which contribute to aging [52–54]. Notably, β-gal staining is a marker of hyperfunctional lysosomes [55]. A combination of markers, including mTOR targets, is needed to define senescence [33]. Some senolytics that target Bcl2 family proteins may theoretically kill leukemia/lymphoma cells. I hope to discuss these and other issues in a scheduled review “Senolytics, gerosuppressants and conventional life-extending drugs.”
Conflicts of Interest
The author declares no conflicts of interest.
References
- 1. Kirkland JL, Tchkonia T. Clinical strategies and animal models for developing senolytic agents. Exp Gerontol. 2015; 68:19–25. https://doi.org/10.1016/j.exger.2014.10.012 [PubMed]
- 2. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O’Hara SP, LaRusso NF, Miller JD, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015; 14:644–58. https://doi.org/10.1111/acel.12344 [PubMed]
- 3. Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc. 2017; 65:2297–301. https://doi.org/10.1111/jgs.14969 [PubMed]
- 4. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018; 24:1246–56. https://doi.org/10.1038/s41591-018-0092-9 [PubMed]
- 5. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016; 22:78–83. https://doi.org/10.1038/nm.4010 [PubMed]
- 6. Wang Y, Chang J, Liu X, Zhang X, Zhang S, Zhang X, Zhou D, Zheng G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY). 2016; 8:2915–26. https://doi.org/10.18632/aging.101100 [PubMed]
- 7. Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, Grassi D, Gregg SQ, Stripay JL, Dorronsoro A, Corbo L, Tang P, Bukata C, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017; 8:422. https://doi.org/10.1038/s41467-017-00314-z [PubMed]
- 8. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, Wren JD, Niedernhofer LJ, Robbins PD, Kirkland JL. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016; 15:428–35. https://doi.org/10.1111/acel.12445 [PubMed]
- 9. Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T, Kirkland JL. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY). 2017; 9:955–63. https://doi.org/10.18632/aging.101202 [PubMed]
- 10. Grezella C, Fernandez-Rebollo E, Franzen J, Ventura Ferreira MS, Beier F, Wagner W. Effects of senolytic drugs on human mesenchymal stromal cells. Stem Cell Res Ther. 2018; 9:108. https://doi.org/10.1186/s13287-018-0857-6 [PubMed]
- 11. Hwang HV, Tran DT, Rebuffatti MN, Li CS, Knowlton AA. Investigation of quercetin and hyperoside as senolytics in adult human endothelial cells. PLoS One. 2018; 13:e0190374. https://doi.org/10.1371/journal.pone.0190374 [PubMed]
- 12. Mikawa R, Suzuki Y, Baskoro H, Kanayama K, Sugimoto K, Sato T, Sugimoto M. Elimination of p19ARF -expressing cells protects against pulmonary emphysema in mice. Aging Cell. 2018; 17:e12827. https://doi.org/10.1111/acel.12827 [PubMed]
- 13. Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner M, Wagner DE, Günther A, Königshoff M. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo.. Eur Respir J. 2017; 50:1602367. https://doi.org/10.1183/13993003.02367-2016 [PubMed]
- 14. Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, Lin S, Huang L, Chung EJ, Citrin DE, Wang Y, Hauer-Jensen M, Zhou D, Meng A. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int J Radiat Oncol Biol Phys. 2017; 99:353–61. https://doi.org/10.1016/j.ijrobp.2017.02.216 [PubMed]
- 15. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017; 8:14532. https://doi.org/10.1038/ncomms14532 [PubMed]
- 16. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016; 354:472–77. https://doi.org/10.1126/science.aaf6659 [PubMed]
- 17. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016; 15:973–77. https://doi.org/10.1111/acel.12458 [PubMed]
- 18. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 2017; 23:1072–79. https://doi.org/10.1038/nm.4385 [PubMed]
- 19. Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, van Deursen JM, Campisi J, Elisseeff JH. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017; 23:775–81. https://doi.org/10.1038/nm.4324 [PubMed]
- 20. Xu M, Bradley EW, Weivoda MM, Hwang SM, Pirtskhalava T, Decklever T, Curran GL, Ogrodnik M, Jurk D, Johnson KO, Lowe V, Tchkonia T, Westendorf JJ, Kirkland JL. Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J Gerontol A Biol Sci Med Sci. 2017; 72:780–85. [PubMed]
- 21. Valentijn FA, Falke LL, Nguyen TQ, Goldschmeding R. Cellular senescence in the aging and diseased kidney. J Cell Commun Signal. 2018; 12:69–82. https://doi.org/10.1007/s12079-017-0434-2 [PubMed]
- 22. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, Grellscheid SN, Hoeijmakers JH, Barnhoorn S, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017; 8:15691. https://doi.org/10.1038/ncomms15691 [PubMed]
- 23. Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018; 562:578–82. https://doi.org/10.1038/s41586-018-0543-y [PubMed]
- 24. Schafer MJ, Haak AJ, Tschumperlin DJ, LeBrasseur NK. Targeting Senescent Cells in Fibrosis: Pathology, Paradox, and Practical Considerations. Curr Rheumatol Rep. 2018; 20:3. https://doi.org/10.1007/s11926-018-0712-x [PubMed]
- 25. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5:2087–102. https://doi.org/10.4161/cc.5.18.3288 [PubMed]
- 26. Blagosklonny MV. TOR-driven aging: speeding car without brakes. Cell Cycle. 2009; 8:4055–59. https://doi.org/10.4161/cc.8.24.10310 [PubMed]
- 27. Gems D, de la Guardia Y. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. 2013; 19:321–29. https://doi.org/10.1089/ars.2012.4840 [PubMed]
- 28. Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013; 75:621–44. https://doi.org/10.1146/annurev-physiol-030212-183712 [PubMed]
- 29. de la Guardia Y, Gilliat AF, Hellberg J, Rennert P, Cabreiro F, Gems D. Run-on of germline apoptosis promotes gonad senescence in C. elegans. Oncotarget. 2016; 7:39082–96. https://doi.org/10.18632/oncotarget.9681 [PubMed]
- 30. Dhondt I, Petyuk VA, Cai H, Vandemeulebroucke L, Vierstraete A, Smith RD, Depuydt G, Braeckman BP. FOXO/DAF-16 Activation Slows Down Turnover of the Majority of Proteins in C. elegans. Cell Reports. 2016; 16:3028–40. https://doi.org/10.1016/j.celrep.2016.07.088 [PubMed]
- 31. Wang H, Zhao Y, Ezcurra M, Benedetto A, Gilliat AF, Hellberg J, Ren Z, Galimov ER, Athigapanich T, Girstmair J, Telford MJ, Dolphin CT, Zhang Z, Gems D. A parthenogenetic quasi-program causes teratoma-like tumors during aging in wild-type C. elegans.. NPJ Aging Mech Dis. 2018; 4:6. https://doi.org/10.1038/s41514-018-0025-3 [PubMed]
- 32. Scialò F, Sriram A, Naudí A, Ayala V, Jové M, Pamplona R, Sanz A. Target of rapamycin activation predicts lifespan in fruit flies. Cell Cycle. 2015; 14:2949–58. https://doi.org/10.1080/15384101.2015.1071745 [PubMed]
- 33. Blagosklonny MV. Rapamycin, proliferation and geroconversion to senescence. Cell Cycle. 2018; 1–11. https://doi.org/10.1080/15384101.2018.1554781 [PubMed]
- 34. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011; 30:1536–48. https://doi.org/10.1038/emboj.2011.69 [PubMed]
- 35. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015; 17:1049–61. https://doi.org/10.1038/ncb3195 [PubMed]
- 36. Christy B, Demaria M, Campisi J, Huang J, Jones D, Dodds SG, Williams C, Hubbard G, Livi CB, Gao X, Weintraub S, Curiel T, Sharp ZD, Hasty P. p53 and rapamycin are additive. Oncotarget. 2015; 6:15802–13. https://doi.org/10.18632/oncotarget.4602 [PubMed]
- 37. Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, Fixler Y, Shreibman D, Zamir A, et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med. 2016; 22:412–20. https://doi.org/10.1038/nm.4054 [PubMed]
- 38. Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol. 2014; 220:T1–23. https://doi.org/10.1530/JOE-13-0584 [PubMed]
- 39. Extermann M. Metabolic syndrome, aging, and cancer. Crit Rev Oncog. 2013; 18:515–29. https://doi.org/10.1615/CritRevOncog.2014010612 [PubMed]
- 40. Blagosklonny MV. TOR-centric view on insulin resistance and diabetic complications: perspective for endocrinologists and gerontologists. Cell Death Dis. 2013; 4:e964. https://doi.org/10.1038/cddis.2013.506 [PubMed]
- 41. Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009; 8:1896–900. https://doi.org/10.4161/cc.8.12.8809 [PubMed]
- 42. Kochetkova EY, Blinova GI, Bystrova OA, Martynova MG, Pospelov VA, Pospelova TV. Targeted elimination of senescent Ras-transformed cells by suppression of MEK/ERK pathway. Aging (Albany NY). 2017; 9:2352–75. https://doi.org/10.18632/aging.101325 [PubMed]
- 43. Kochetkova EY, Blinova GI, Bystrova OA, Martynova MG, Pospelov VA, Pospelova TV. Suppression of mTORC1 activity in senescent Ras-transformed cells neither restores autophagy nor abrogates apoptotic death caused by inhibition of MEK/ERK kinases. Aging (Albany NY). 2018; 10:3574–89. https://doi.org/10.18632/aging.101686 [PubMed]
- 44. Slack C, Alic N, Partridge L. Could cancer drugs provide ammunition against aging? Cell Cycle. 2016; 15:153–55. https://doi.org/10.1080/15384101.2015.1118905 [PubMed]
- 45. Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?”. Aging (Albany NY). 2012; 4:861–77. https://doi.org/10.18632/aging.100525 [PubMed]
- 46. Velarde MC, Demaria M, Campisi J. Senescent cells and their secretory phenotype as targets for cancer therapy. Interdiscip Top Gerontol. 2013; 38:17–27. https://doi.org/10.1159/000343572 [PubMed]
- 47. Blagosklonny MV. Prevention of cancer by inhibiting aging. Cancer Biol Ther. 2008; 7:1520–24. https://doi.org/10.4161/cbt.7.10.6663 [PubMed]
- 48. Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY). 2011; 3:1130–41. https://doi.org/10.18632/aging.100422 [PubMed]
- 49. Blagosklonny MV. Selective anti-cancer agents as anti-aging drugs. Cancer Biol Ther. 2013; 14:1092–97. https://doi.org/10.4161/cbt.27350 [PubMed]
- 50. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012; 4:159–65. https://doi.org/10.18632/aging.100443 [PubMed]
- 51. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7:3344–54. https://doi.org/10.4161/cc.7.21.6965 [PubMed]
- 52. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, Leonova K, Polinsky A, Chernova OB, Gudkov AV. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY). 2016; 8:1294–315. https://doi.org/10.18632/aging.100991 [PubMed]
- 53. Frescas D, Hall BM, Strom E, Virtuoso LP, Gupta M, Gleiberman AS, Rydkina E, Balan V, Vujcic S, Chernova OB, Gudkov AV. Murine mesenchymal cells that express elevated levels of the CDK inhibitor p16(Ink4a) in vivo are not necessarily senescent. Cell Cycle. 2017; 16:1526–33. https://doi.org/10.1080/15384101.2017.1339850 [PubMed]
- 54. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin II, Leonova KI, Consiglio CR, Gollnick SO, et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY). 2017; 9:1867–84. https://doi.org/10.18632/aging.101268 [PubMed]
- 55. Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000; 113:3613–22. [PubMed]