



Introduction
Cellular senescence is defined as the irreversible arrest of cell proliferation. In the 1960s, Hayflick et al. found that normal human diploid fibroblasts entered into an irreversible non-dividing state after a certain number of divisions, which was referred to as “Hayflick limit” [1]. Since then, multiple types of cellular senescence have been identified including replicative senescence, oncogene-induced senescence, DNA damage-induced senescence, oxidative stress-induced senescence, chemotherapy-induced senescence, mitochondrial dysfunction-associated senescence, epigenetically induced senescence, paracrine senescence, wound healing and embryonic development related senescence [2]. However, whether all of those types of senescence model occur in vivo still remains unknown.
In general, senescent cells are characterized by the enlarged cell size, increased lysosomal content and upregulated β-galactosidase activity at nearly pH 7.0 [3]. Cellular senescence is established and maintained by at least two major tumor suppressor pathways [4], the p53/p21 and the p16Ink4a/retinoblastoma protein (Rb) axes. It is believed that the p53/p21 axis initiates the senescence process, while the p16Ink4a activation maintains the senescence state [5]. In cultured cells, senescence occurs as a defensive mechanism to resolve cellular insults, leading to transient cell cycle arrest. In this case, cells can re-enter cell cycle once the stress is resolved. Prolonged cellular stress (> 4 days), however, spurs permanent senescence [6]. Other than ceased cell division, senescent cells also display widespread changes in chromatin structure (referred to as senescence associated heterochromatin foci, SAHF) and gene expression profiles [7], which synergistically lead to highly active cellular metabolism and massive secretion of cytokines (TGF-β, IL-1a, -1β and -6), chemokines (IL-8, CXCL1), growth factors (FGF, HGF) and proteases (MMP-1, -3, and -13), collectively defined as senescence associated secretory phenotypes (SASP) [3, 8]. Interestingly, senescent cells manifest loss of Lamin B1 expression, but the related mechanism and significance are yet to be explored [9]. SASP is a characteristic feature shared by almost all senescent cells, and it is mainly initiated by the NF-κB and p38MAPK pathways, while maintained by IL-1α in an autocrine manner [10]. The composition of the senescence-associated secretome varies depending on the time spent in senescence, the senescence inducer and the cell type [11]. Two main distinct secretomes have been described, and the NOTCH1 signaling plays a pivotal role in switching secretome composition [12]. During the early stage of senescence, NOTCH1 activity fluctuates dynamically, which triggers a TGF-β rich secretome to suppresses the senescence-associated pro-inflammatory secretome by inhibiting C/EBPβ signaling. However, sustained senescence endows NOTCH1-driven TGF-β to repress NOTCH1 signaling transduction, which in turn contributes to the second wave of senescence induction, thereby changing the TGF-β rich secretome into a pro-inflammatory-centered one [12, 13].
It is believed that senescence represents a programmed phenomenon that facilitates mammalian embryonic development and β cell functional maturation after birth [14, 15]. A couple of senescence hallmarks including p16Ink4a, p19Arf and p15Ink4b increase in pancreatic β cells during aging, along with decreased capability of regeneration [16–18]. A large body of work has focused on the impact of senescent β cell accumulation on the pathogenesis of type 1 diabetes (T1D) and progression of age-related type 2 diabetes (T2D) [19, 20]. These studies open up new perspectives to understand aging and diabetes development, which would promote the exploitation of promising therapeutic strategies. In this review, we summarize the recent progress in aging-, and stress-induced β cell senescence, and its impact on β cell viability, insulin secretion and regeneration, as well as discuss its relevance to the development of diabetes mellitus.
Characteristics of β cell senescence
Studies in rodents and humans have revealed that recovery and plasticity of islet cells decrease in mice once they reached 1-year of age, and human β cell population is established by the age of 20 [21] except for the existence of a small population of “virgin β cell”, which is functionally immature [22]. Those facts are reminiscent of natural β cell senescence with age. Generally, senescent β cells exhibit larger cell size (~14um) than the normal (~12um), and can be featured by the upregulated expression of Cdkn2a/1a (encoding p16Ink4a and p21, respectively) and anti-apoptotic molecules (e.g., Bcl-2, -xl and -w) along with senescence associated β-galactosidase staining [19, 20]. Specific composition of senescence-associated secretome helps to distinguish β cell senescence with senescence in other cell types. Of note, distinctive features and signatures of β cell senescence exist in T1D and T2D disease models in multiple ways, indicating that β cell senescence is dynamically regulated under different cellular contexts.
Aging and stress (e.g., hyperglycemia, viral insult, inflammatory response and insulin resistance) can be contributors to β cell senescence. Studies have demonstrated that aging causes massive changes of β cell chromatin accessibility, leading to significant alterations in gene expression profile [23]. Nevertheless, the association of senescence phenotype with β cell epigenetic and transcriptomic changes during aging still need further exploration. Indeed, single cell RNA-seq analysis demonstrated that aged human pancreas manifests enhanced transcriptional noise, somatic mutations and senescence signatures [24]. Emerging evidence shows that sources and levels of DNA damage increase with age along with decreased DNA repair capacity [25], predisposing β cells to cell cycle arrest and DNA damage response (DDR) associated with senescence. Cell replication-related telomere erosion is known to directly associate with lifespan limitation [26]. There are data supporting that short telomere impairs β cell function and participates in β cell destruction in the late stage of T2D [27]. Furthermore, proteomic analysis reveals that β cells manifest a significant discrepancy in terms of the expression of aging markers (e.g., IGF1R) between islets even within the same islets, suggesting that β cells display a remarkable aging heterogeneity [28]. Hyperglycemia is another trigger of β cell senescence. β cells maintain blood glucose homeostasis by controlling appropriate insulin secretion according to the real-time changes of blood glucose levels [29]. Sustained hyperglycemia, however, would induce β cell senescence via multiple possible mechanisms, such as apoptosis signaling-regulating kinase 1 activation [30], p38 mitogen-activated protein kinase activation [31], and “glycolytic overload” (characterized by the increased metabolic flux through glycolysis in hyperglycemia and the decreased proteolysis of hexokinase) mediated mitochondrial dysfunction [32]. Importantly, unlike other cell types, β cells manifest relatively lower antioxidant capability and, as a result, they are more susceptible to oxidative stress and endoplasmic reticulum (ER) stress [33–35]. Excessive reactive oxygen species (ROS) production impairs mitochondrial dynamics (fission and fusion), leading to defective electron transport chain, bioenergetics imbalance, and altered mitochondria calcium homeostasis, which then trigger β cell senescence [36–38]. It is noteworthy that mitochondria-related senescence is featured by the lack of IL1-arm cytokines, due to high AMP to ATP ratio in the mitochondria coupled with highly activated AMPK, which in turn represses the initiation of mTORC1 and IL1-arm cytokine responses [39]. This feature, however, has not been validated in β cells.
Increased protein synthesis load, oxidative stress, gene mutations, glucolipotoxicity, can cause ER tress in β cells. The activation of the three branches of unfolded protein response accelerates cellular senescence in non-β cell types, and the ER chaperone, Bip, has a possible central role in senescence [38, 40]. Therefore, it is quite possible that ER stress actively participates in β cell senescence despite the unclear molecular mechanisms. Virus, especially enteroviruses, is one of the origins of β cell DNA damage [41]. Indeed, recent studies demonstrated that islets with infiltrated immune cells are characterized by the increased frequency of DDR and enhanced expression of senescence markers in newly diagnosed T1D patients and rodent T1D model [42], indicating that DNA damage-induced β cell senescence may play a critical role in the early stage of autoimmune diabetes. Up to now, however, the contribution of virus insult to β cell senescence and T1D progression remains to be described. Nevertheless, the existing data support that autoimmune response in T1D and chronic inflammation in T2D are suspect culprits of β cell senescence, possibly through ER stress, DNA damage and other signaling pathways. While systemic insulin resistance accelerates β cell senescence during aging, the involved molecular mechanisms, however, are not fully understood. Collectively, during the course of aging and diabetes progression, multiple triggers and signaling pathways collaborate and twist together to induce β cell senescence, leading to changes in β cell functions and systemic metabolic homeostasis in cell-autonomous and noncell-autonomous manners.
The effect of senescence on β cell regeneration
β cell cycling is driven by CyclinD1/2-CDK4 activity and downregulated by CDK inhibitor p16Ink4a. It has proven that β cell expansion is an age-dependent process, and β cell replication is much more robust in young mice than that in old animals [43]. Once p16Ink4a is specifically expressed, pancreatic β cells show obvious senescence phenotype along with compromised cell regeneration [17]. Since p16Ink4a transcript is enriched in purified islets when compared with exocrine tissues, supporting that p16Ink4a serves as a crucial checkpoint in β cell senescence as well as proliferation. Conversely, p16Ink4a ablation enhances β cell proliferation, especially in the case of β cells following toxic insult [16]. It has been shown that two chromatin-regulating polycomb group proteins, Bmi1 and Ezh2, are related to age dependent high levels of p16Ink4a expression in β cells, suggesting that epigenetic regulation could be involved in senescence-mediated aging and type 2 diabetes [44, 45]. Indeed, mice deficient in pituitary tumor transforming gene (PTTG), which encodes a securing protein that regulates chromosome separation, go through evident senescence and apoptosis in islet β cells at 2-month-old [46]. Enhanced p21 expression in β cells following PTTG knockout could be one of the contributors to senescent phenotype, because p21 deletion only partially rescued mice from diabetes resulted from severe β cell diminishment. Those discoveries support the notion that β cell senescence can be secondary to DNA damage associated gene activation, and furthermore, additional PTTG downstream genes may also synergize with p21 attributing to β cell senescence and cycling arrest.
To develop the strategies for β cell expansion against aging, a great deal of research effort has been focused on the regulation of β cell regeneration/senescence. Platelet-derived growth factor (PDGFR) signaling has been characterized to play a critical role in cellular proliferation and development. PDGFR losses its expression with age both in mouse and human islet β cells along with deceased EZH2 expression [45]. Conditional over-activation of PDGFRa in β cells enhances neonatal β cell propagation and regeneration in adult islets [47]. Juvenile human islets rather than adult islets exposed to PDGF-AA, a PDGF-A agonist, rejuvenate β cell proliferation. Similarly, exendin-4, an agonist of the glucagon like peptide 1 receptor (GLP-1R), successfully stimulates juvenile human β cell expansion but fails in adult islet β cells [48]. These findings are expected, because aging process causes overall changes of chromatin accessibility and gene expression profile toward the activation of metabolic regulator and suppression of proliferation program, which is a reminiscent of irreversibility of normal aging (Figure 1).
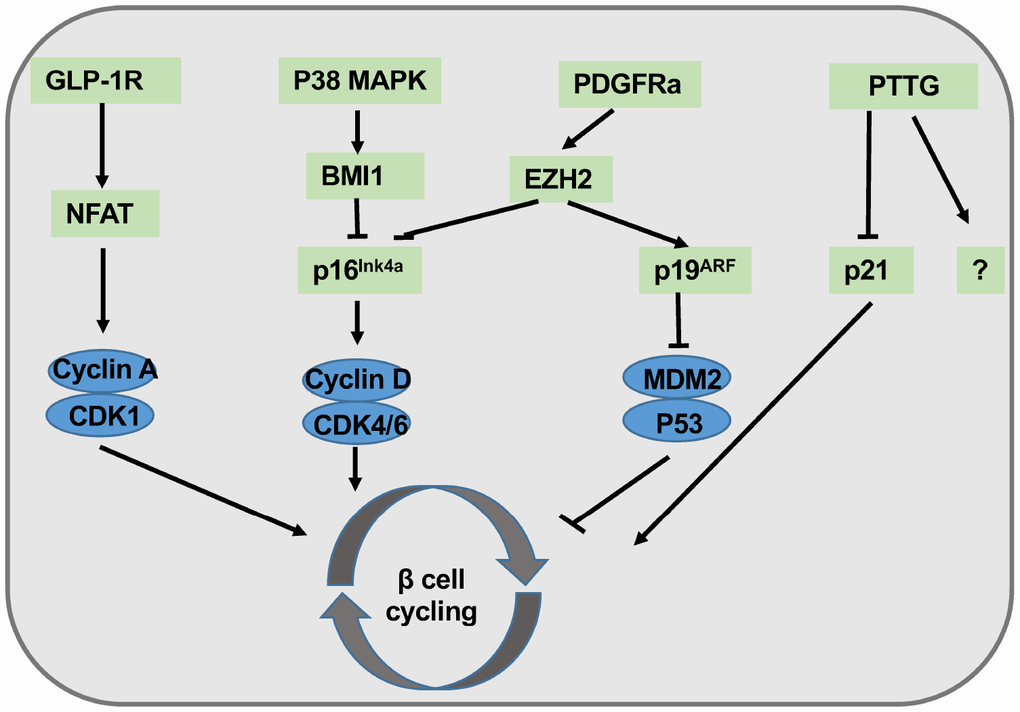
Figure 1. Summary of molecular pathways involved in β cell regeneration. Exendin-4 agonizes GLP-1R signaling, followed by the activation of NFAT and the entry of cell cycling. P38 MAPK signals activate BMI1 and inhibit p16Ink4a. PDGFRa transduces proliferative signals to EZH2, thereby attenuating p16Ink4a activity while enhancing p19Arf activation. PTTG partially promotes β cell proliferation via p21 inhibition.
The influences of β cell senescence in insulin secretion
The implication of aging in the regulation of insulin synthesis/secretion and glucose homeostasis had been recognized back to 1980s. By utilizing Fischer rat, an established aging animal model, Wang etal. found that aging has no effect on preproinsulin mRNA transcription, but impairs nearly half amount of proinsulin synthesis upon high glucose stimulation [49]. Undoubtedly, decreased proinsulin synthesis would lead to the reduction of newly formed insulin secretion. Given that pancreatic weight, total insulin content, islet size and mean insulin content per islet are unchanged, the impairments in the signal transduction following glucose stimulation during aging process could be a crucial culprit. Indeed, studies with time-dependent potentiation (TDP) of insulin release in aged rats confirmed that β cells lose sensitivity to secretagogues during aging process [50]. However, the exact mechanisms had not been dissected at that time. Actually, in a β cell specific p16Ink4a overexpression mouse model, ectopic p16Ink4a expression improved glucose stimulated insulin secretion response apart from cell cycle arresting [17]. Although this finding is consistent with previous data demonstrating that mitochondrial metabolism and insulin exocytosis relevant to β cell functions are improved during aging process, but contradicts to other findings [23, 49, 50]. The following possible reasons may explain the above discrepancies. On one hand, transgenic p16Ink4a expression in β cell may only simulate one facet of cellular senescence to partially reflect β cell senescence phenotype, but sustained decrease of β cell function in elderly individuals cannot be neglected [50]. On the other hand, insulin synthesis and secretion in aged subjects are likely modulated by multiple factors such as senescence marker protein-30 (SMP-30), an androgen independent factor involving in Vitamin C synthesis that decreases during aging process to impair GSIS in elders [51]. Furthermore, elevated plasma level of deoxysphingolipid is responsible for the senescent characteristics and compromised GSIS both in INS-1 cells and primary islets [52]. Another noteworthy phenomenon is that senescent β cells manifest higher basal insulin level (2.8mM glucose), which is similar to the immature β cell phenotype. This puzzle may be partially explained by NAD(P)H fluorescence lifetime imaging (FLIM) implication. Aging causes β cell mitochondria dysfunction mainly through complex I/II disorder followed by reduction of KATP channel activity and increase of Ca2+ inflowing that occur as a compensatory strategy, thereby increasing insulin exocytosis [53].
More recently, studies indicated that β cell senescence can be affected by proximal pancreatic cells namely acinar cells and other hormonal factors. It was noted that the expression of arginase II in acinar cells increases during aging process, and enhanced TNF-α release from acinar cells induces β cell dysfunction and apoptosis [38]. Since β cell senescence can be disseminated by surrounding β cells, a crosstalk may exist between β cells and other types of pancreatic cells, thereby regulating β cell senescence. Indeed, thyroid hormone (T3) spurs β cell functional maturation through MafA induction, and enhances cell senescence by directly activating its target p16Ink4a [54] through TH receptor B (THRB) and TH receptor A (THRA). Arum et al. demonstrated that mice deficient in growth hormone receptor (GHR)/binding protein gene display hypoinsulinemia, higher sensitivity to insulin and prolonged lifespan even though they are smaller in size [55], while insertion of Igf1 gene under the control of a rat insulin promoter (RIP) can reverse the above phenotypes, supporting that IGF1R is a new aging marker [28, 55]. Consistently, transgenic Igf1 expression in conventional GHR knockout mice highlights that insulin sensitivity is important for longevity, and the pace of aging can actually be hormonally regulated [56]. (Figure 2)
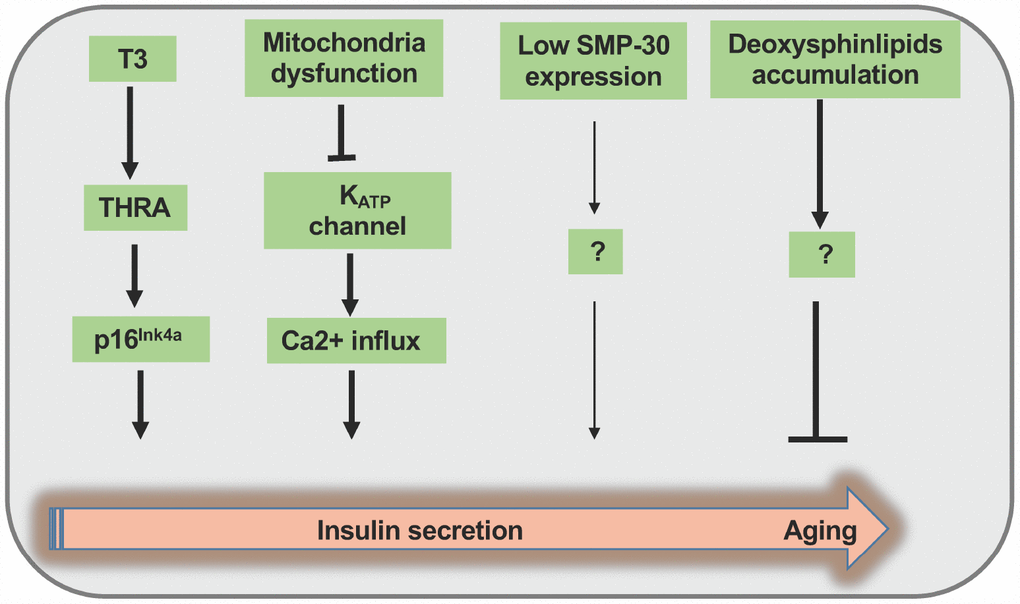
Figure 2. Summary of the regulation of insulin secretion during the course of aging process. Thyroid hormone (T3) promotes β cell functional maturation via the induction of MafA expression along with p16Ink4a activation at the early stage of aging. During the progression of aging process, impaired mitochondria function causes KATP channel shutting down and Ca2+ influx at the same time, thereby enhancing insulin secretion in a short time frame. In contrast, during the advanced aging process, β cells manifest diminished expression of senescence marker protein-30 (SMP-30) along with deoxysphingolipid accumulation, thereby impeding insulin secretion through unknown mechanisms.
It would be necessary to keep in mind that several essential questions need to be fully addressed to dissect the association between senescence and β cell function thoroughly. First, how does senescence regulate proinsulin synthesis in β cells? second, how many glucose responding proteins are subject to β cell senescence? and third, how does the enhanced basal insulin secretion occur and what is the relevance to aging or diabetes mellitus?
The relevance of β cell senescence to diabetes development
T1D is featured by the progressive β cell destruction from autoimmune response. Recently, Thompson et al. demonstrated that a subpopulation of senescent β cells exists in both NOD mice and human T1D patients, and they actively recruit autoimmune cells, indicating a pivotal role of β cell senescence in T1D progression [20]. Specifically, in T1D-prone NOD mice, the autoimmune cells initiate peri-insulitis (the recruitment of autoreactive immune cells into the periphery of islets) with minor β cell destruction from 3-4 weeks to 8-10 weeks, while the disease progresses after 10 weeks, which causes invasive insulitis accompanied by the massive β cell destruction and severe hyperglycemia [57]. The single β cell transcription profile comparison between the two stages of T1D reveals that senescent β cells accumulate with disease progression, exhibit DNA damage and stress-induced senescence phenotypes. The senescent cells are conspicuous due to the upregulated senescent markers consist of p16Ink4a, p21, Ser139-phosphorylated histone H2A.X (γ-H2A.X), the increased SASP markers including Cxcl10, IL-6, Mmp-2 and Flnb, as well as specific secretome (including IL-6, Igfbp3 and Serpine1). Notably, the SASP factors potently enforce the paracrine effect of senescence and the chemotaxis of immune cells. When translating those findings to human T1D cases, senescent human β cells display some distinctive characteristics. Firstly, in human β cells, p16Ink4a is more likely to be an age-related senescence marker rather a T1D-related one. In contrast, p21 expression drastically increases in autoantibody-positive nondiabetic donors and newly diagnosed T1D donors, which renders p21 to be a human T1D-related senescence marker. Secondly, the senescent secretome in human T1D is featured by IL-6 and Serpine1 expression. Lastly, heterogeneity among islets and individuals is obvious in terms of the aforementioned senescent marker expression levels. The senescent NOD β cells highly express Bcl-2, but the anti-apoptotic feature in human senescent β cells, however, remains undefined due to unknown limitations. While human T1D onset can take a couple of years or decades, stress-induced, instead of age-related senescence, take the primary responsibility for disease progression. There is strong evidence that autonomous β cell DDR along with the presence of the hallmarks of senescence can play a causal role in autoimmune initiation and progression during the course of T1D development [42]. However, β cell DNA damage caused by autoimmunity does not involve in this process as the major senescent β cell in NOD islets are not surrounded by immune cells.
T2D is an aging-associated disease characterized by the systemic insulin resistance and metabolic dysfunction in multiple organs and tissues, which includes two stages, the pre-diabetes stage and the early-stage diabetes. The pre-diabetes stage can sustain for many years attributing to β cell compensation (i.e., increased β cell mass and workload) till β cell compensation failure, which leads to the second stage featured by β cell death, decreased insulin levels in the circulation and prolonged hyperglycemia [58]. The boundary of the two stages, however, is difficult to define since most patients remain in a grey area of diagnosis, where diet changes and antidiabetic drugs are sufficient to maintain normoglycemia. The connection between β cell senescence and T2D has been established for several years [59]. To understand the effects of β cell senescence on T2D pathogenesis, aging and stress factors including hyperglycemia, hyperlipidemia and chronic inflammation, should be taken into account. Aged β cells accumulate with age, which exhibit impaired insulin secretion response to glucose challenges. It was noted that β cells in aged C57BL/6 mice display a distinctive transcription profile characterized by the downregulation of β cell identity genes (including Insulin1, MafA, Nkx6.1, and Pdx1), upregulation of senescence markers (including Cav1, Cdkn1a, Id2, Makp2k1, Prkcd, Tbx2 and Tbx3), SASP genes (e.g., Ccl2, Cd68, Igfbp3, Il6, Tnf and Serpine1) and disallowed genes that are supposed to be repressed in β cells, such as Ldha and Catalase [19, 60]. Nevertheless, those mouse-derived data are only partially consistent with what found in aged human islets [28]. It is noteworthy that insulin resistance can exacerbate the senescent process in aged β cells [19]. However, unlike high-fat-diet induced senescence, β cell senescence induced by acute administration of S961, an insulin receptor antagonist, can be reversed once S961 is withdrawal [61]. Despite the unclear mechanisms, this finding indicates that β cell senescence can be reversed at early stage, and therefore, the impact of β cell senescence on clinical T2D progression could be much more complicated than what we thought. Indeed, in humans, the proportion of senescent β cells (characterized by the upregulation of CXCL10, CCL4,IL1A and IL6) substantially increased in aged subjects, and further increased in subjects with T2D. However, only two SASP factors (CCL4 and IL6) are detected in human T2D patients, left the impact of β cell senescence on human T2D progression an unfinished story [62]. Nevertheless, insulin resistance has been consistently found to be tightly associated with hyperglycemia, hyperlipidemia and chronic inflammation both in T2D patients and animals, and those factors are linked to the occurrence of β cell senescent phenotypes and SASP activities. In the pre-diabetes stage, insulin-resistance-related high blood glucose, dyslipidemia and inflammation increase insulin demand, impelling the β cell expansion to secret more insulin [14]. β cell adaptive regeneration shortens telomere and activates DDR, followed by cellular senescence. On the other hand, paracrine senescence accelerates senescent β cell accumulation and promotes SASP activities, which in turn exacerbate systemic insulin resistance and enhance the loss of β cell compensation, coupled with the progression of pre-diabetes to the early diabetes stage. However, it would be difficult to define in which diabetes stage that β cell senescence is more important, because of the vague boundary of the two stages and the prolonged timeframe of β cell senescence.
Recently, Dooley et al. identified β cell variations in genes Xrcc4 and Glis3 in the NOD islets, both of which share links to T2D susceptibility [63, 64], supporting genetic predisposition of β cell senescence in diabetes risk [65]. Furthermore, SNPs adjacent to the CDKN2a/b gene have been identified and attested to associate with T2D in large GWAS studies [66]. These discoveries highlight that β cell senescence related genetic defects may increase the susceptibility of T1D and T2D. Collectively, β cell senescence is a common contributor to T1D and aging-related T2D. It seems that DDR is a common trigger of β cell senescence both in T1D and aging-associated T2D. However, whether this pathway is the most essential one to drive β cell senescence and diabetes onset remains to be further explored. Obviously, distinctive origins of β cell senescence delineate different senescence signatures and secretomes, suggesting distinctive senescent mechanisms and SASP effects on T1D and T2D. Since cellular senescence is a dynamic process varying with cell type, senescence inducer and time of duration, those properties in β cell senescence, however, have not been fully described in present studies.
Concluding remarks and future directions
Cellular senescence is certainly crucial to growth and development at early stage of life. Specifically, senescence promotes β cell functional maturation including increased glucose uptake, mitochondrial oxidation capability and mitochondria number. However, sustained senescence is associated with aging-related lifespan limitation and disease development. β cell senescence during aging impairs the expression of genes relevant to β cell identity and cellular functions. Furthermore, SASP-derived cytokines could trigger inflammatory response, which renders β cell status even worse. Although the established mouse model with p16Ink4a overexpression in around 35% of β cells largely resembles the phenotypes observed in normal aging mice, but some discrepancies are noted. For example, aged mice show impaired response to glucose fluctuations, whereas p16Ink4a overexpression merely improves high glucose stimulated insulin secretion. Therefore, cellular senescence is a more complicated entity involving multiple molecules and signaling pathways [9]. As such, p16Ink4a overexpressing cells just manifest a part of senescence phenotype, but lack of other upstream/downstream signals and cellular alterations. Particularly, aged human islets display an age dependent decline in the coordination of Ca2+ dynamics, gap junction coupling and insulin secretion [67], and p16Ink4a mediated improvement of GSIS does not seem to be durable, because deteriorated glucose tolerance is noted once induced p16Ink4a expression lasts for 5-month.
Studies in a genetic senescence activation mouse model revealed that prolonged β cell senescence deteriorates cellular function followed by β cell exhaustion and β cell death no matter what type of cell death it is [17]. Furthermore, during the course of natural aging process, islet cells from both aged human and rat are sensitive to glucose induced β cell apoptosis confirmed by TUNEL staining [68]. Given that senescent cells are resistant to apoptosis, the effect of senescent cells is mostly dependent on their SASP activities including paracrine senescence and chemotaxis, which may explain β cell destruction and decreased β cell mass associated hyperglycemia. A critical question is whether targeted clearance of senescent cells would attenuate diabetes development. To address this question, large number of pharmacological compounds (defined as “senolytics”) have been identified to specifically induce senescent cell death [2]. Those senolytic drugs are aimed at combating some chronic diseases (e.g., diabetes, neurodegenerative diseases) or extending lifespan, even though with possible unclear side effects [69–74]. In this case, the effect of various senolytic chemicals have been exploited intensively on multiple disease models including T1D and T2D [19, 20, 75]. Specifically, senolytic induction of senescent β cells by two BH3 mimetics, ABT-737 (inhibits Bcl-2, Bcl-xl and Bcl-w) [76] and ABT-199 (a FDA-approved drug which inhibits Bcl-2 specifically) [77], effectively halts T1D development in NOD mice [20]. Similarly, ABT-263 reverses T2D outcomes by improving β cell function and identity [19, 78]. In consideration of the pros and cons of these senolytic drugs on trial, next generations of potent senolytic compounds targeting diabetes with minor side effects should be developed urgently based on β cell senescence and SASP properties. Encouragingly, several approved antidiabetic drugs actually exhibit anti-senescence effect to some extents. For example, metformin has now been tested in the Targeting Aging with Metformin trials as a good regimen against aging and age-related diseases [79–82]. Since calorie restriction improves longevity [83, 84], and metformin treatment just mimics dietary restriction, which highly upregulates AMPK activity while inhibits mTORC1 activity and NF-κB pathway, thereby modulating autophagy/senescence and ameliorating cardiovascular diseases [85, 86]. It has been noted that chronic low dose metformin treatment extends lifespan by increasing the nuclear accumulation of nuclear factor erythroid 2-related factor 2 (Nrf2) to facilitate an array of expressions for antioxidant genes [60]. Additionally, dipeptidyl-peptidase 4 inhibitor and rosiglitazone also display plausible effects on ameliorating senescence in non-β cell types [87], unveiling novel molecular mechanisms in antidiabetic therapies.
At present, several questions are still remained to be investigated in terms of the role of β cell senescence in diabetes pathogenesis. First, as β cell senescence has been associated with both T1D and T2D, detailed mechanistic studies are necessary to explain how β cell senescence affects diabetes development (e.g., the role of β cell senescence in the initiation and progression of diabetes). Second, previous studies suggested links between senescence and autophagy [88, 89] and cell reprogramming [90–92], but whether those links can also apply to β cells is yet to be clarified, especially in diabetic context. Third, the dynamic property of β cell senescence and senescence associated secretome still needs further exploration. Lastly, extensive assessments for the future of senolytic therapies against diabetes are still necessary before its application in clinical settings. In conclusion, we have summarized the recent progress in β cell senescence, through which we intend to spark more instructive discussion and perspective with regard to the mechanisms underlying β cell senescence and their links to the pathogenesis of diabetes and the development of therapeutic strategies. We believe that a comprehensive understanding of β cell senescence would provide great potential to the prevention and treatment of diabetes.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
Our research is supported by the National Natural Science Foundation of China (81530024, 91749207, 81471046, 81770823 81470988, 81920108009 and 81670929), the Ministry of Science and Technology (2016YFC1305002 and 2017YFC1309603), NHC Drug Discovery Program (2017ZX09304022-07), the Department of Science and Technology of Hubei State (2017ACA096), the Integrated Innovative Team for Major Human Disease Programs of Tongji Medical College, Huazhong University of Science and Technology, and the Innovative Funding for Translational Research from Tongji Hospital.
References
- 1. Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965; 37:614–36. https://doi.org/10.1016/0014-4827(65)90211-9 [PubMed]
- 2. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018; 28:436–53. https://doi.org/10.1016/j.tcb.2018.02.001 [PubMed]
- 3. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015; 15:397–408. https://doi.org/10.1038/nrc3960 [PubMed]
- 4. Martínez-Zamudio RI, Robinson L, Roux PF, Bischof O. SnapShot: Cellular Senescence Pathways. Cell. 2017; 170:816–816.e1. https://doi.org/10.1016/j.cell.2017.07.049 [PubMed]
- 5. Stein GH, Drullinger LF, Soulard A, Dulić V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999; 19:2109–17. https://doi.org/10.1128/MCB.19.3.2109 [PubMed]
- 6. Chen X, Zhang W, Gao YF, Su XQ, Zhai ZH. Senescence-like changes induced by expression of p21(waf1/Cip1) in NIH3T3 cell line. Cell Res. 2002; 12:229–33. https://doi.org/10.1038/sj.cr.7290129 [PubMed]
- 7. Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, Thuret JY, Andrews S, Fraser P, Reik W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015; 10:471–83. https://doi.org/10.1016/j.celrep.2014.12.055 [PubMed]
- 8. Newgard CB, Sharpless NE. Coming of age: molecular drivers of aging and therapeutic opportunities. J Clin Invest. 2013; 123:946–50. https://doi.org/10.1172/JCI68833 [PubMed]
- 9. He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017; 169:1000–11. https://doi.org/10.1016/j.cell.2017.05.015 [PubMed]
- 10. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011; 25:2125–36. https://doi.org/10.1101/gad.17276711 [PubMed]
- 11. Schmitt CA. The persistent dynamic secrets of senescence. Nat Cell Biol. 2016; 18:913–15. https://doi.org/10.1038/ncb3403 [PubMed]
- 12. Hoare M, Ito Y, Kang TW, Weekes MP, Matheson NJ, Patten DA, Shetty S, Parry AJ, Menon S, Salama R, Antrobus R, Tomimatsu K, Howat W, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. 2016; 18:979–92. https://doi.org/10.1038/ncb3397 [PubMed]
- 13. Boni A, Urbanek K, Nascimbene A, Hosoda T, Zheng H, Delucchi F, Amano K, Gonzalez A, Vitale S, Ojaimi C, Rizzi R, Bolli R, Yutzey KE, et al. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci USA. 2008; 105:15529–34. https://doi.org/10.1073/pnas.0808357105 [PubMed]
- 14. Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, Spitale RC, Dai C, Gu X, Qu K, Wang P, Wang J, Grompe M, et al. Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human β Cell Function. Cell Metab. 2016; 23:909–20. https://doi.org/10.1016/j.cmet.2016.04.002 [PubMed]
- 15. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M. Programmed cell senescence during mammalian embryonic development. Cell. 2013; 155:1104–18. https://doi.org/10.1016/j.cell.2013.10.019 [PubMed]
- 16. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006; 443:453–57. https://doi.org/10.1038/nature05092 [PubMed]
- 17. Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, Fixler Y, Shreibman D, Zamir A, et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med. 2016; 22:412–20. https://doi.org/10.1038/nm.4054 [PubMed]
- 18. Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009; 23:906–11. https://doi.org/10.1101/gad.1742609 [PubMed]
- 19. Aguayo-Mazzucato C, Andle J, Lee TB
Jr , Midha A, Talemal L, Chipashvili V, Hollister-Lock J, van Deursen J, Weir G, Bonner-Weir S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019; 30:129–142.e4. https://doi.org/10.1016/j.cmet.2019.05.006 [PubMed] - 20. Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019; 29:1045–1060.e10. https://doi.org/10.1016/j.cmet.2019.01.021 [PubMed]
- 21. Cnop M, Igoillo-Esteve M, Hughes SJ, Walker JN, Cnop I, Clark A. Longevity of human islet α- and β-cells. Diabetes Obes Metab. 2011 (Suppl 1); 13:39–46. https://doi.org/10.1111/j.1463-1326.2011.01443.x [PubMed]
- 22. van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dólleman S, Liu S, Ackermann AM, Cáceres E, Hunter AE, Kaestner KH, Donaldson CJ, Huising MO. Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab. 2017; 25:911–926.e6. https://doi.org/10.1016/j.cmet.2017.03.017 [PubMed]
- 23. Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, Stadler MB, Burger L, Schübeler D, Glaser B, Kaestner KH. Aging-Dependent Demethylation of Regulatory Elements Correlates with Chromatin State and Improved β Cell Function. Cell Metab. 2015; 22:619–32. https://doi.org/10.1016/j.cmet.2015.07.025 [PubMed]
- 24. Swisa A, Kaestner KH, Dor Y. Transcriptional Noise and Somatic Mutations in the Aging Pancreas. Cell Metab. 2017; 26:809–11. https://doi.org/10.1016/j.cmet.2017.11.009 [PubMed]
- 25. Niedernhofer LJ, Gurkar AU, Wang Y, Vijg J, Hoeijmakers JH, Robbins PD. Nuclear Genomic Instability and Aging. Annu Rev Biochem. 2018; 87:295–322. https://doi.org/10.1146/annurev-biochem-062917-012239 [PubMed]
- 26. Wątroba M, Dudek I, Skoda M, Stangret A, Rzodkiewicz P, Szukiewicz D. Sirtuins, epigenetics and longevity. Ageing Res Rev. 2017; 40:11–19. https://doi.org/10.1016/j.arr.2017.08.001 [PubMed]
- 27. Guo N, Parry EM, Li LS, Kembou F, Lauder N, Hussain MA, Berggren PO, Armanios M. Short telomeres compromise β-cell signaling and survival. PLoS One. 2011; 6:e17858. https://doi.org/10.1371/journal.pone.0017858 [PubMed]
- 28. Aguayo-Mazzucato C, van Haaren M, Mruk M, Lee TB
Jr , Crawford C, Hollister-Lock J, Sullivan BA, Johnson JW, Ebrahimi A, Dreyfuss JM, Van Deursen J, Weir GC, Bonner-Weir S. β Cell Aging Markers Have Heterogeneous Distribution and Are Induced by Insulin Resistance. Cell Metab. 2017; 25:898–910.e5. https://doi.org/10.1016/j.cmet.2017.03.015 [PubMed] - 29. Semple RK, Williams RM, Dunger DB. What is the best management strategy for patients with severe insulin resistance? Clin Endocrinol (Oxf). 2010; 73:286–90. https://doi.org/10.1111/j.1365-2265.2010.03810.x [PubMed]
- 30. Yokoi T, Fukuo K, Yasuda O, Hotta M, Miyazaki J, Takemura Y, Kawamoto H, Ichijo H, Ogihara T. Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes. 2006; 55:1660–65. https://doi.org/10.2337/db05-1607 [PubMed]
- 31. Kuki S, Imanishi T, Kobayashi K, Matsuo Y, Obana M, Akasaka T. Hyperglycemia accelerated endothelial progenitor cell senescence via the activation of p38 mitogen-activated protein kinase. Circ J. 2006; 70:1076–81. https://doi.org/10.1253/circj.70.1076 [PubMed]
- 32. Rabbani N, Thornalley PJ. Hexokinase-2 Glycolytic Overload in Diabetes and Ischemia-Reperfusion Injury. Trends Endocrinol Metab. 2019; 30:419–31. https://doi.org/10.1016/j.tem.2019.04.011 [PubMed]
- 33. Chakraborty S, Rasool RU, Kumar S, Nayak D, Rah B, Katoch A, Amin H, Ali A, Goswami A. Cristacarpin promotes ER stress-mediated ROS generation leading to premature senescence by activation of p21(waf-1). Age (Dordr). 2016; 38:62. https://doi.org/10.1007/s11357-016-9922-1 [PubMed]
- 34. Drews G, Krippeit-Drews P, Düfer M. Oxidative stress and beta-cell dysfunction. Pflugers Arch. 2010; 460:703–18. https://doi.org/10.1007/s00424-010-0862-9 [PubMed]
- 35. Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia. 2013; 56:234–41. https://doi.org/10.1007/s00125-012-2762-3 [PubMed]
- 36. Song M, Franco A, Fleischer JA, Zhang L, Dorn GW
2nd . Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017; 26:872–883.e5. https://doi.org/10.1016/j.cmet.2017.09.023 [PubMed] - 37. Wiley CD, Campisi J. From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab. 2016; 23:1013–21. https://doi.org/10.1016/j.cmet.2016.05.010 [PubMed]
- 38. Kim HS, Kim Y, Lim MJ, Park YG, Park SI, Sohn J. The p38-activated ER stress-ATF6α axis mediates cellular senescence. FASEB J. 2019; 33:2422–34. https://doi.org/10.1096/fj.201800836R [PubMed]
- 39. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016; 23:303–14. https://doi.org/10.1016/j.cmet.2015.11.011 [PubMed]
- 40. Liu Y, Zhu H, Yan X, Gu H, Gu Z, Liu F. Endoplasmic reticulum stress participates in the progress of senescence and apoptosis of osteoarthritis chondrocytes. Biochem Biophys Res Commun. 2017; 491:368–73. https://doi.org/10.1016/j.bbrc.2017.07.094 [PubMed]
- 41. Roivainen M, Rasilainen S, Ylipaasto P, Nissinen R, Ustinov J, Bouwens L, Eizirik DL, Hovi T, Otonkoski T. Mechanisms of coxsackievirus-induced damage to human pancreatic beta-cells. J Clin Endocrinol Metab. 2000; 85:432–40. https://doi.org/10.1210/jcem.85.1.6306 [PubMed]
- 42. Horwitz E, Krogvold L, Zhitomirsky S, Swisa A, Fischman M, Lax T, Dahan T, Hurvitz N, Weinberg-Corem N, Klochendler A, Powers AC, Brissova M, Jörns A, et al. β-Cell DNA Damage Response Promotes Islet Inflammation in Type 1 Diabetes. Diabetes. 2018; 67:2305–18. https://doi.org/10.2337/db17-1006 [PubMed]
- 43. Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005; 54:2557–67. https://doi.org/10.2337/diabetes.54.9.2557 [PubMed]
- 44. Gilbert ER, Liu D. Epigenetics: the missing link to understanding β-cell dysfunction in the pathogenesis of type 2 diabetes. Epigenetics. 2012; 7:841–52. https://doi.org/10.4161/epi.21238 [PubMed]
- 45. Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, Kim SK. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009; 23:975–85. https://doi.org/10.1101/gad.1742509 [PubMed]
- 46. Chesnokova V, Wong C, Zonis S, Gruszka A, Wawrowsky K, Ren SG, Benshlomo A, Yu R. Diminished pancreatic beta-cell mass in securin-null mice is caused by beta-cell apoptosis and senescence. Endocrinology. 2009; 150:2603–10. https://doi.org/10.1210/en.2008-0972 [PubMed]
- 47. Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature. 2011; 478:349–55. https://doi.org/10.1038/nature10502 [PubMed]
- 48. Dai C, Hang Y, Shostak A, Poffenberger G, Hart N, Prasad N, Phillips N, Levy SE, Greiner DL, Shultz LD, Bottino R, Kim SK, Powers AC. Age-dependent human β cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J Clin Invest. 2017; 127:3835–44. https://doi.org/10.1172/JCI91761 [PubMed]
- 49. Wang SY, Halban PA, Rowe JW. Effects of aging on insulin synthesis and secretion. Differential effects on preproinsulin messenger RNA levels, proinsulin biosynthesis, and secretion of newly made and preformed insulin in the rat. J Clin Invest. 1988; 81:176–84. https://doi.org/10.1172/JCI113291 [PubMed]
- 50. Bombara M, Masiello P, Novelli M, Bergamini E. Impairment of the priming effect of glucose on insulin secretion from isolated islets of aging rats. Acta Diabetol. 1995; 32:69–73. https://doi.org/10.1007/BF00581050 [PubMed]
- 51. Hasegawa G, Yamasaki M, Kadono M, Tanaka M, Asano M, Senmaru T, Kondo Y, Fukui M, Obayashi H, Maruyama N, Nakamura N, Ishigami A. Senescence marker protein-30/gluconolactonase deletion worsens glucose tolerance through impairment of acute insulin secretion. Endocrinology. 2010; 151:529–36. https://doi.org/10.1210/en.2009-1163 [PubMed]
- 52. Zuellig RA, Hornemann T, Othman A, Hehl AB, Bode H, Güntert T, Ogunshola OO, Saponara E, Grabliauskaite K, Jang JH, Ungethuem U, Wei Y, von Eckardstein A, et al. Deoxysphingolipids, novel biomarkers for type 2 diabetes, are cytotoxic for insulin-producing cells. Diabetes. 2014; 63:1326–39. https://doi.org/10.2337/db13-1042 [PubMed]
- 53. Gregg T, Poudel C, Schmidt BA, Dhillon RS, Sdao SM, Truchan NA, Baar EL, Fernandez LA, Denu JM, Eliceiri KW, Rogers JD, Kimple ME, Lamming DW, Merrins MJ. Pancreatic β-Cells From Mice Offset Age-Associated Mitochondrial Deficiency With Reduced KATP Channel Activity. Diabetes. 2016; 65:2700–10. https://doi.org/10.2337/db16-0432 [PubMed]
- 54. Aguayo-Mazzucato C, Lee TB
Jr , Matzko M, DiIenno A, Rezanejad H, Ramadoss P, Scanlan T, Zavacki AM, Larsen PR, Hollenberg A, Colton C, Sharma A, Bonner-Weir S. T3 Induces Both Markers of Maturation and Aging in Pancreatic β-Cells. Diabetes. 2018; 67:1322–31. https://doi.org/10.2337/db18-0030 [PubMed] - 55. Arum O, Boparai RK, Saleh JK, Wang F, Dirks AL, Turner JG, Kopchick JJ, Liu JL, Khardori RK, Bartke A. Specific suppression of insulin sensitivity in growth hormone receptor gene-disrupted (GHR-KO) mice attenuates phenotypic features of slow aging. Aging Cell. 2014; 13:981–1000. https://doi.org/10.1111/acel.12262 [PubMed]
- 56. Zhao X. SUMO-Mediated Regulation of Nuclear Functions and Signaling Processes. Mol Cell. 2018; 71:409–18. https://doi.org/10.1016/j.molcel.2018.07.027 [PubMed]
- 57. Graham KL, Sutherland RM, Mannering SI, Zhao Y, Chee J, Krishnamurthy B, Thomas HE, Lew AM, Kay TW. Pathogenic mechanisms in type 1 diabetes: the islet is both target and driver of disease. Rev Diabet Stud. 2012; 9:148–68. https://doi.org/10.1900/RDS.2012.9.148 [PubMed]
- 58. Heianza Y, Arase Y, Fujihara K, Tsuji H, Saito K, Hsieh SD, Kodama S, Shimano H, Yamada N, Hara S, Sone H. Screening for pre-diabetes to predict future diabetes using various cut-off points for HbA(1c) and impaired fasting glucose: the Toranomon Hospital Health Management Center Study 4 (TOPICS 4). Diabet Med. 2012; 29:e279–85. https://doi.org/10.1111/j.1464-5491.2012.03686.x [PubMed]
- 59. Fang J, Yang J, Wu X, Zhang G, Li T, Wang X, Zhang H, Wang CC, Liu GH, Wang L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell. 2018; 17:e12765. https://doi.org/10.1111/acel.12765 [PubMed]
- 60. Lemaire K, Thorrez L, Schuit F. Disallowed and Allowed Gene Expression: Two Faces of Mature Islet Beta Cells. Annu Rev Nutr. 2016; 36:45–71. https://doi.org/10.1146/annurev-nutr-071715-050808 [PubMed]
- 61. Schäffer L, Brand CL, Hansen BF, Ribel U, Shaw AC, Slaaby R, Sturis J. A novel high-affinity peptide antagonist to the insulin receptor. Biochem Biophys Res Commun. 2008; 376:380–83. https://doi.org/10.1016/j.bbrc.2008.08.151 [PubMed]
- 62. Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. 2005; 48:58–67. https://doi.org/10.1007/s00125-004-1605-2 [PubMed]
- 63. Fortune MD, Guo H, Burren O, Schofield E, Walker NM, Ban M, Sawcer SJ, Bowes J, Worthington J, Barton A, Eyre S, Todd JA, Wallace C. Statistical colocalization of genetic risk variants for related autoimmune diseases in the context of common controls. Nat Genet. 2015; 47:839–46. https://doi.org/10.1038/ng.3330 [PubMed]
- 64. Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, Horikoshi M, Johnson AD, Ng MC, Prokopenko I, Saleheen D, Wang X, Zeggini E, et al, and DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, and Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, and South Asian Type 2 Diabetes (SAT2D) Consortium, and Mexican American Type 2 Diabetes (MAT2D) Consortium, and Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014; 46:234–44. https://doi.org/10.1038/ng.2897 [PubMed]
- 65. Dooley J, Tian L, Schonefeldt S, Delghingaro-Augusto V, Garcia-Perez JE, Pasciuto E, Di Marino D, Carr EJ, Oskolkov N, Lyssenko V, Franckaert D, Lagou V, Overbergh L, et al. Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nat Genet. 2016; 48:519–27. https://doi.org/10.1038/ng.3531 [PubMed]
- 66. Bao XY, Xie C, Yang MS. Association between type 2 diabetes and CDKN2A/B: a meta-analysis study. Mol Biol Rep. 2012; 39:1609–16. https://doi.org/10.1007/s11033-011-0900-5 [PubMed]
- 67. Westacott MJ, Farnsworth NL, St Clair JR, Poffenberger G, Heintz A, Ludin NW, Hart NJ, Powers AC, Benninger RK. Age-Dependent Decline in the Coordinated [Ca2+] and Insulin Secretory Dynamics in Human Pancreatic Islets. Diabetes. 2017; 66:2436–45. https://doi.org/10.2337/db17-0137 [PubMed]
- 68. Maedler K, Schumann DM, Schulthess F, Oberholzer J, Bosco D, Berney T, Donath MY. Aging correlates with decreased beta-cell proliferative capacity and enhanced sensitivity to apoptosis: a potential role for Fas and pancreatic duodenal homeobox-1. Diabetes. 2006; 55:2455–62. https://doi.org/10.2337/db05-1586 [PubMed]
- 69. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell. 2017; 169:132–147.e16. https://doi.org/10.1016/j.cell.2017.02.031 [PubMed]
- 70. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016; 530:184–89. https://doi.org/10.1038/nature16932 [PubMed]
- 71. Serrano M. Ageing: tools to eliminate senescent cells. Nature. 2017; 545:294–96. https://doi.org/10.1038/nature22493 [PubMed]
- 72. Knoppert SN, Valentijn FA, Nguyen TQ, Goldschmeding R, Falke LL. Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front Pharmacol. 2019; 10:770. https://doi.org/10.3389/fphar.2019.00770 [PubMed]
- 73. Hampel H, Lista S, Neri C, Vergallo A, Alzheimer Precision Medicine I. Time for the systems-level integration of aging: resilience enhancing strategies to prevent Alzheimer’s disease. Prog Neurobiol. 2019; 181:101662. https://doi.org/10.1016/j.pneurobio.2019.101662 [PubMed]
- 74. Papatheodoridi AM, Chrysavgis L, Koutsilieris M, Chatzigeorgiou A. The role of senescence in the development of non-alcoholic fatty liver disease and progression to non-alcoholic steatohepatitis. Hepatology. 2019. [Epub ahead of print]. https://doi.org/10.1002/hep.30834 [PubMed]
- 75. Gurău F, Baldoni S, Prattichizzo F, Espinosa E, Amenta F, Procopio AD, Albertini MC, Bonafè M, Olivieri F. Anti-senescence compounds: A potential nutraceutical approach to healthy aging. Ageing Res Rev. 2018; 46:14–31. https://doi.org/10.1016/j.arr.2018.05.001 [PubMed]
- 76. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005; 435:677–81. https://doi.org/10.1038/nature03579 [PubMed]
- 77. Davids MS, Letai A. ABT-199: taking dead aim at BCL-2. Cancer Cell. 2013; 23:139–41. https://doi.org/10.1016/j.ccr.2013.01.018 [PubMed]
- 78. Park CM, Bruncko M, Adickes J, Bauch J, Ding H, Kunzer A, Marsh KC, Nimmer P, Shoemaker AR, Song X, Tahir SK, Tse C, Wang X, et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem. 2008; 51:6902–15. https://doi.org/10.1021/jm800669s [PubMed]
- 79. Arunachalam G, Samuel SM, Marei I, Ding H, Triggle CR. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br J Pharmacol. 2014; 171:523–35. https://doi.org/10.1111/bph.12496 [PubMed]
- 80. Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017; 16:718–35. https://doi.org/10.1038/nrd.2017.116 [PubMed]
- 81. Forouzandeh F, Salazar G, Patrushev N, Xiong S, Hilenski L, Fei B, Alexander RW. Metformin beyond diabetes: pleiotropic benefits of metformin in attenuation of atherosclerosis. J Am Heart Assoc. 2014; 3:e001202. https://doi.org/10.1161/JAHA.114.001202 [PubMed]
- 82. Geiger H. Depleting senescent cells to combat aging. Nat Med. 2016; 22:23–24. https://doi.org/10.1038/nm.4024 [PubMed]
- 83. Fontana L, Nehme J, Demaria M. Caloric restriction and cellular senescence. Mech Ageing Dev. 2018; 176:19–23. https://doi.org/10.1016/j.mad.2018.10.005 [PubMed]
- 84. Zhang N, Li Z, Mu W, Li L, Liang Y, Lu M, Wang Z, Qiu Y, Wang Z. Calorie restriction-induced SIRT6 activation delays aging by suppressing NF-κB signaling. Cell Cycle. 2016; 15:1009–18. https://doi.org/10.1080/15384101.2016.1152427 [PubMed]
- 85. Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN, Ferbeyre G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell. 2013; 12:489–98. https://doi.org/10.1111/acel.12075 [PubMed]
- 86. Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, Laboy R, Hirschey MD, Mair WB. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab. 2017; 26:884–896.e5. https://doi.org/10.1016/j.cmet.2017.09.024 [PubMed]
- 87. Chen L, Bi B, Zeng J, Zhou Y, Yang P, Guo Y, Zhu J, Yang Q, Zhu N, Liu T. Rosiglitazone ameliorates senescence-like phenotypes in a cellular photoaging model. J Dermatol Sci. 2015; 77:173–81. https://doi.org/10.1016/j.jdermsci.2015.01.007 [PubMed]
- 88. Cassidy LD, Narita M. CELL BIOLOGY. GATA get a hold on senescence. Science. 2015; 349:1448–49. https://doi.org/10.1126/science.aad2501 [PubMed]
- 89. Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ, Puleston DJ, Watson AS, Simon AK, Tooze SA, Akbar AN. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8⁺ T cells. J Clin Invest. 2014; 124:4004–16. https://doi.org/10.1172/JCI75051 [PubMed]
- 90. Alfego D, Rodeck U, Kriete A. Global mapping of transcription factor motifs in human aging. PLoS One. 2018; 13:e0190457. https://doi.org/10.1371/journal.pone.0190457 [PubMed]
- 91. Mosteiro L, Pantoja C, de Martino A, Serrano M. Senescence promotes in vivo reprogramming through p16INK4a and IL-6. Aging Cell. 2018; 17:e12711. https://doi.org/10.1111/acel.12711 [PubMed]
- 92. Milanovic M, Fan DN, Belenki D, Däbritz JH, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, Mendoza-Parra MA, Kanashova T, Metzner M, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018; 553:96–100. https://doi.org/10.1038/nature25167 [PubMed]