



Introduction
Nowadays, stroke has become the leading cause of adult disability and the second most prominent cause of death worldwide, only after coronary heart disease. Ischemic stroke almost accounts for 80% in all stroke cases [1]. Over the past decades, the established therapeutic option for ischemic stroke patients is still limited to recanalization of occlusive vessels with the clot-breaking agent tissue plasminogen activator (t-PA). However, due to the serious tissue damage which may occur during the subsequent reperfusion (such as bleeding) and the limited therapeutic time window (within 4.5h post stroke), more than 90% of ischemic stroke patients are unavailable to intravenous t-PA therapy [2]. Although numerous potential pathophysiologic mechanisms and targets for ischemic stroke have been found in recent years, they are rarely translated into feasible medical practice [3].
Tau is a protein mainly expressed in the brain, it has six isoforms produced by alternative mRNA splicing of microtubule-associated protein tau (MAPT) gene which comprises 16 exons on chromosome 17q21 [4] (Figure 1A). The primary physiological function of tau protein is to stabilize microtubule networks within neurons, whereas the hyperphosphorylated condition will significantly reduce its biological activity [5]. Although previous studies mainly focused on the mechanisms of tau protein in neurodegenerative diseases [6, 7], some studies have also demonstrated that increased tau immunoreactivity after brain ischemia-reperfusion injury can be observed in neuronal cells [8, 9]. Recently, several novel functions of tau protein have been revealed [10, 11]. Whereas the association between tau protein and ischemic stroke has not been well discussed. In this review, we aim to update the knowledge about the genomic and proteomic changes in tau protein following ischemia/reperfusion injury and the connection between tau protein and ischemic stroke.
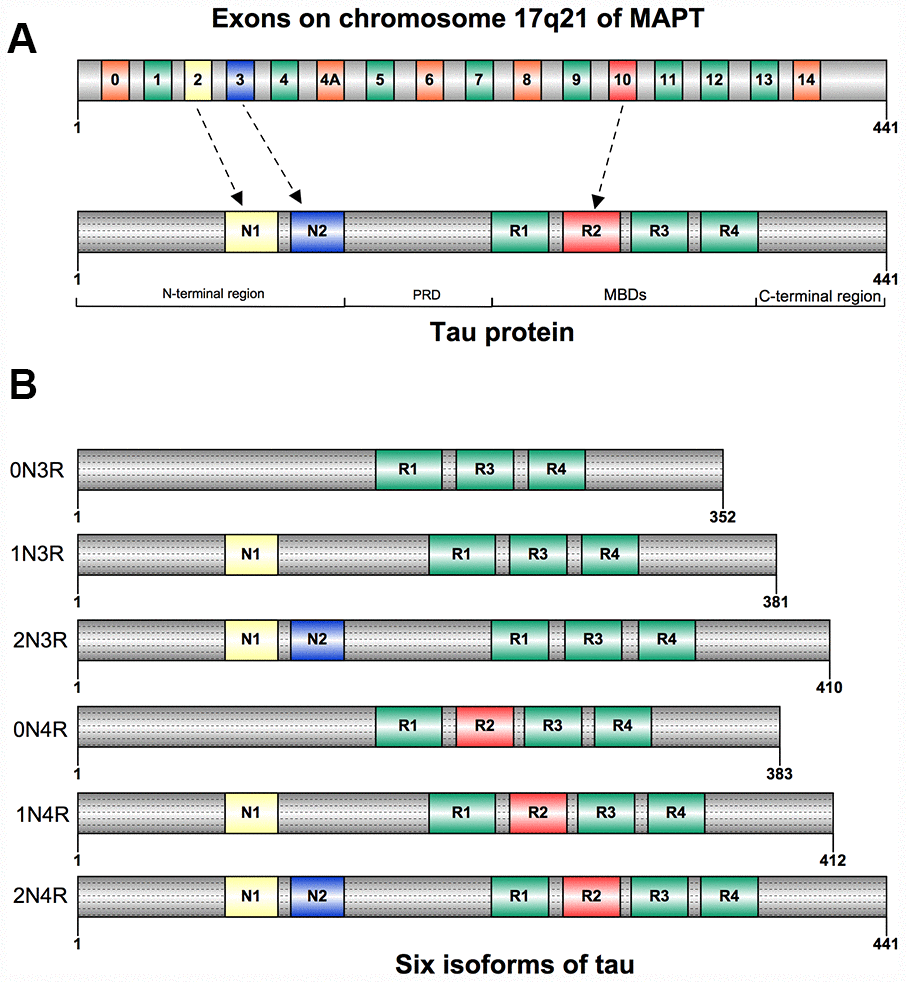
Figure 1. (A) Structure of human tau protein; Tau has an N-terminao projection region, a proline-rich domain(PRD), a microtubule-binding domain(MBD), and a C-terminal region. (B) Six isoforms of human tau. They differ by the inclusion of exon 2(NI), exon 3(N2), and exonlO(RI-R4).
Structure and biological functions of tau
Tau was first isolated and named in 1975 for its ability to induce tubule formation [12], and was mostly segregated into neuronal axons [13]. Tau can be also detected in oligodendrocytes and neuronal somatodendritic compartments [14]. Besides the nervous system, tau was also found to be expressed in many other tissues: heart, lung, kidney, and testis, but less abundant [15]. Tau is composed of four regions: an N-terminal projection region, a proline-rich domain (PRD), a microtubule-binding domain (MBD), and a C-terminal region [16] (Figure 1A). Six isoforms of tau have been found in human adult brain, they are expressed by alternative splicing around the N-terminal projection region and MBD. The gene expression of these isoforms differs both in N-terminal exons (0N, 1N, or 2N) and the number of microtubule binding repeat sequences (3R or 4R). The 4R tau has four microtubule binding repeat sequences due to the inclusion of exon 10 when compared with 3R tau [4, 17] (Figure 1B).
The mainly physiological tau function in the cell is regulating microtubule structure and dynamics by binding to microtubules, it has been also proven in cell-free conditions [12]. Furthermore, the dynamic microtubule network provided by tau is key to the proper migration of new neurons, and severe reduction of adult neurogenesis was found in tau knockout mice [18]. Tau also plays an important role in controlling the balance of microtubule-dependent axonal transport through the differential sensitivity of motor proteins in neurons [19]. Additionally, it has been approved that tau is essential in the protection of neuronal genomic DNA and RNA integrity under hyperthermia condition both in primary neuronal cultures and in vivo in adult mice [20]. Besides, absence or reduction of tau expression has been reported to have protective efforts against memory deficits, excitotoxicity, amyloid induced toxicity, and epilepsy in animal experiments [21, 22] (Figure 2).
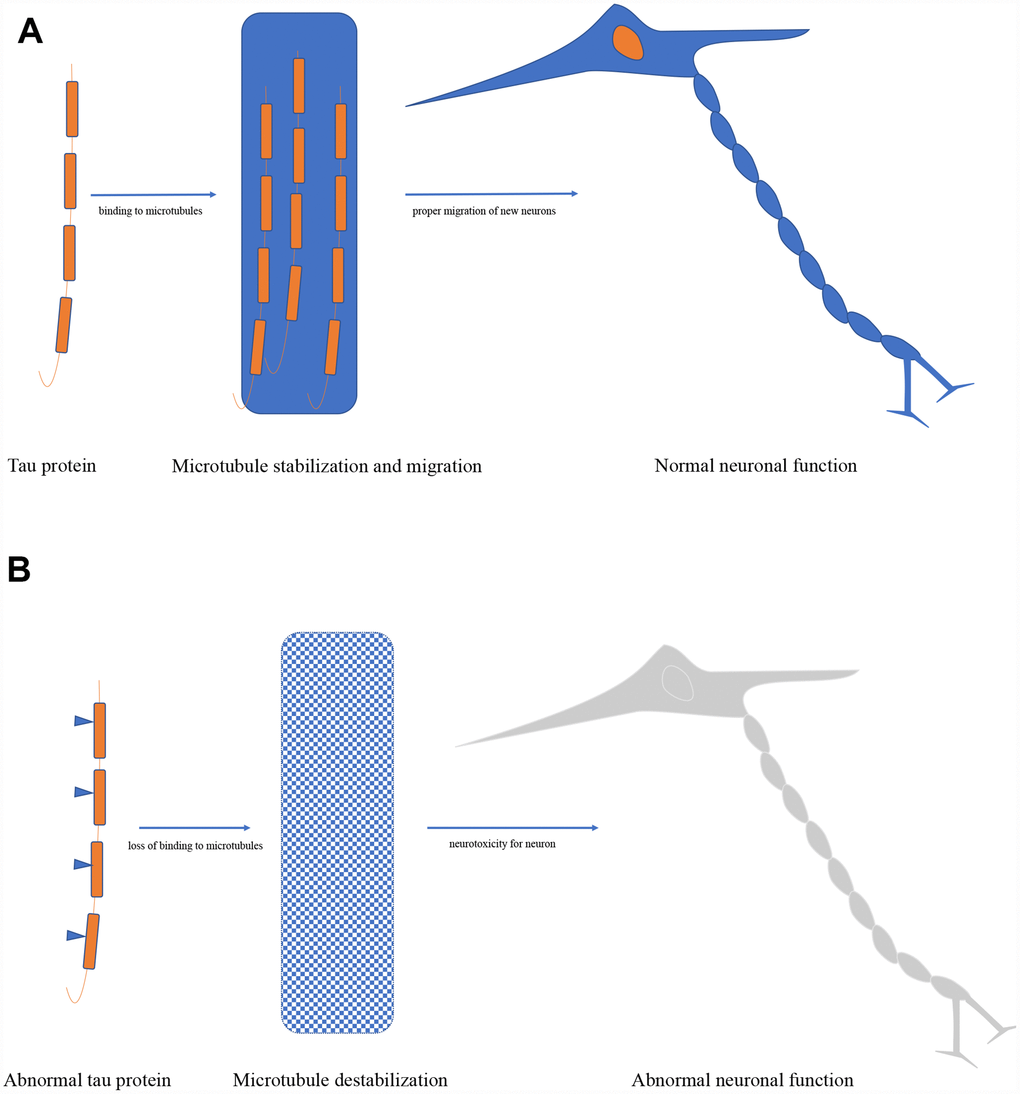
Figure 2. (A) Biological function of tau protein. (B) Pathological role of tau protein.
Potential mechanisms of tau in ischemic stroke
Tau functions are regulated by a complex array of post-translational modifications, such as phosphorylation, glycation, acetylation, isomerization, nitration, sumoylation, O-GlcNAcylation, and truncation [16, 23], suggesting that tau plays diverse roles in physiology and pathology. Dysfunctional tau is one of the neurotoxic proteins, accumulated in neurons and cerebrovascular after ischemia, furthermore, it is closely related to a range of pathological changes of ischemic stroke [24, 25]. According to previous studies, the kinds of dysfunctional tau differ in different ischemic models, such as neurofibrillary tangle formation [26–28], hyperphosphorylation [29–34], dephosphorylation [8, 35–39], and re-phosphorylation [8, 40] (Table 1). The hyperphosphorylated state is the particularly pathological condition of tau in brain ischemia. It decreases the affinity of tau for the microtubules by disrupting the binding balance [5, 30–34, 41]. In this part, we will summarize the potential regulatory mechanisms of tau in ischemic stroke.
Table 1. Patterns of Tau Phosphorylation in Brain after Ischemic Stroke
| References | Human/Animal | Models/Subjects | Ischemic time | Analyzed tissue | State of tau protein | Tau phospho-sites | Effects of tau |
| Bi M 2017 [11] | Mice | Focal cerebral ischemia model | 90min/ 30min | The cortex in the ischemic area | Tau | N | Reduce tau protein-dependent excitotoxicity in tau–/– mice |
| Basurto IG 2018 [117] | Mice | Focal cerebral ischemia model | 1 hour | The ischemic core | Hyperphosphorylation | Ser262/356 | Hyperphosphorylation involving asparagine endopeptidase |
| Khan S 2018 [27] | Mice | Global cerebral ischemia model | 10,15,18min | The hippocampus and the cortex | Paired helical filament tau protein increase | Ps396/404 | Lead to neuronal death |
| Liao G 2009 [118] | Mice | Right common carotid artery was occluded and hypoxia was maintained | 40 min | The ischemic core | A marked decrease in tau phosphorylation | P301L | Extracellular glutamate accumulation |
| Tuo QZ 2017 [10] | Mice/ Rats | Focal cerebral ischemia model | Mice:60min Rats:90min | The lesioned hemisphere | Tau | N | Dysfunctional or absent tau protein contributes to iron-mediated neurotoxicity |
| Dewar D 1995 [36] | Rats | Focal cerebral ischemia model | 2-6hours | The cortex in the ischemic area | Dephosphorylated and/or degraded | Tau 1 | Breakdown of the cytoskeleton in ischemic region of the neuron |
| Geddes JW 1994 [37] | Rats | Complete cerebral ischemia model | 20 min | The hippocampal formation | Dephosphorylated | Tau 1 | Compromises the ability of the neuron to remove Elevated intracellular Ca2+ |
| Shackelford DA,1998 [39] | Rats | Complete cerebral ischemia model | 5-15min | The hippocampus, neocortex and striatum | Dephosphorylated | Ps396/404 | Possibly contributing to disruption of axonal transport |
| Wen Y 2004 [31] | Rats | Focal cerebral ischemia model | 1 hour | The cortex in the ischemic area | Hyperphosphorylation | PT181, pS202, pT205, pT212, pS214, pT231, pS262, pS396, pS404, and pS422 | Destabilize neuronal cytoskeleton, and may contribute to the Apoptotic process |
| Wen Y 2004 [33] | Rats | Focal cerebral ischemia model | 1 hour | The cortex in the ischemic area | Hyperphosphorylation | MC1 and TG3 (phospho-tau 231/ 235); phosphorylated tau epitopes: CP13 (phospho- tau 202/205), CP3 (phospho-tau 214), PHF-1 (phospho-tau 396/ 404), and CP9 (phospho-tau 231) | Involved in the progression of Neuropathology in AD |
| Kovalska M 2018 [34] | Rats | Global cerebral ischemia model | 15min | The cortex in the ischemic area | Hyperphosphorylation | Ser202, Thr205 | Degeneration of cortical neurons, alterations in number and morphology of tissue astrocytes and dysregulation of Oxidative balance |
| Fujii H 2017 [30] | Rats | Focal cerebral ischemia model | 90 mins | The ischemic core | Hyperphosphorylation | Asp421-truncated tau | Influence microtubule stability and Subsequently disturb axonal transport, resulting in the formation of axonal varicosities and other axonal abnormalities |
| Wen Y 2007 [29] | Rats | Focal cerebral ischemia model | 1 hour | The cortex in the ischemic area | Hyperphosphorylation and neurofibrillary tangle (NFT) like conformations | P-396/404 | Involved in the progression of neuropathology in AD |
| Majd S 2016 [38] | Rats | Global cerebral ischemia model | 8 mins | Parietal cortical and subcortical hippocampus homogenates | Phosphorylation/ dephosphorylation | Ser(396) and Ser(262), Ser(202) /Thr(205) (AT8) | Dephosphorylation of AMPK followed the same pattern as tau dephosphorylation during ischemia or reperfusion |
| Whitehead SN,2005 [28] | Rats | Subcortical Lacunar infarcts by striatal endothelin injections | N | Hippocampus | Neurofibrillary tangles and senile plaques to form | Tau 2 | Mediating neurotoxic and neuroinflammatory |
| Morioka M 2006 [32] | Gerbils | Global forebrain ischemia model | 5 mins | Hippocampal region | Hyperphosphorylation | Serine 199/202 | Induced by MAP kinase, CDK5, and GSK3, and contributes to ischemic neuronal injury |
| Gordon KW 2007 [8] | Gerbils | Global forebrain ischemia model | 5 mins | The cortex in the ischemic area | Hyperphosphorylation | Tau 1 | May caused by oxidative stress |
| Mailliot C 2000 [40] | Dogs | Cardiac arrest -induced global cerebral ischemia | 10mins | The ischemic core | Dephosphorylation, differential and re-phosphorylation | Ser262/356 | Monitor neuronal integrity after brain ischemia |
| Burkhart KK 1998 [35] | Rats/Human | Complete cerebral ischemia model Neocortical brain slices | 5mins/ 30mins | The cortex in the ischemic area | Dephosphorylation and an apparent recovery in phosphorylated tau | Tau 1 | Dephosphorylated tau may enhance Microtubule stability |
| Uchihara T 2004 [127] | Human | Ischemic stroke | N | The cortex in the ischemic area | Hyperphosphorylation | Ser101 | Microglia tau protein passes independent of phosphorylation modification |
| Kato T 1988 [26] | Human | Ischemic stroke | N | The cortex in the ischemic area | Neurofibrillary tangle formation | Tau 1 | These cases may represent an initial stage of senile changes |
Tau and oxidative stress
Oxidative stress is a pathological condition which constitutes the mechanisms of many disease including ischemic stroke. It has been proven in animal experiments that the hyperphosphorylation of tau can be resulted from oxidative stress through different kinds of oxidant, like intracerebroventricular streptozotocin (ICV-STZ) [42, 43], streptozotocin [44] and 1,2- diacetylbenzene (DAB) [45]. On the other hand, hyperphosphorylation of tau can be reduced by antioxidants, such as EUK 207 [46], EUK 189 [47] and exendin-4 (Ex-4) [42]. There is no unified opinion on the underlying mechanisms between oxidative stress and hyperphosphorylation of tau. Many studies have found that polyunsaturated lipids, thiobarbituric acid reactive substances (TBARS), and 4-hydroxynonenal (4-HNE) produced by peroxidation of intracellular lipids are notably increased, which may contribute to hyperphosphorylation of tau [42, 43]. More recently, tau hyperphosphorylation is proven to be directly stimulated by ROS, which is produced by DAB via the phosphorylation of activated glycogen synthase kinase-3β (GSK-3β) [45]. Moreover, high concentration of hyperphosphorylated tau has been shown to stimulate the production of ROS [48]. Therefore, oxidative stress and tau hyperphosphorylation may be two key elements of a vicious circle after ischemic stroke.
Tau and apoptosis
Apoptosis is a dynamically programmed process of cell death, acting an essential actor in the neuronal damage after ischemic stroke [49]. Tau hyperproteolysis/ proteolysis and apoptosis are considered to be two independent pathological events after neuron damage, most researchers did not demonstrate the underlying relationship between them [50, 51]. However, one recent study has proven that the accumulation of CDK5-regulated tau phosphorylation might trigger neuronal apoptosis through impairing endoplasmic reticulum-associated degradation [52]. Researchers also found that tau phosphorylation could be inhibited by knocking down CDK5 (an upstream regulatory factor of tau), which could protect neurons by mitigating endoplasmic reticulum stress from apoptosis [52].
Tau and autophagy
Autophagy is subtyped into constitutive macro-autophagy which plays a major role in maintaining the appropriate levels of functional tau in neurons [53–55]. Autophagy has been indicated to be an important pathophysiological process in both hemorrhagic stroke and ischemic stroke [56, 57]. Previous studies have demonstrated that the decrease in tau is directly correlated with the increase in specific autophagy markers (such as LC3B-II) in the 3xTg-AD mouse model after transient hypoperfusion, indicating that autophagy may be a pathway of lowering dysfunctional tau level after hypoperfusion [58]. Another study has detected a significant decrease in the level of LC3B protein and a reduction in infarct volume in ischemic P301L-Tau mice [59]. The researchers considered it might be possible that the autophagy-mediated degradation is influenced by mutated tau with the increase levels of protein aggregates [59]. Furthermore, it has been demonstrated that regulators of autophagy can mediate tau expression in neurons overexpressing human mutant P301L-Tau [60]. In human tauopathies, p62 is the regulative protein of selective autophagy, and its immunoreactivity co-localizes with tau inclusions [61]. In mice and cells, autophagy activation can promote the clearance of assembled tau [62] and reduce the aggregation of seeded tau [63]. Many studies consider tau phosphorylation a consequence of seeded aggregation [64]. P62 and nuclear dot protein 52 (NDP52) are both autophagy cargo receptors, playing vital role in protecting against seeded tau aggregation in cells [60, 65]. So it is possible that autophagy, rather than the proteasome, restricts the aggregation of seeded tau [60].
Tau and excitotoxicity
Excitotoxicity has been identified as one of the molecular mechanisms of ischemic stroke in many studies [66–68]. Many studies suggest that tau phosphorylation can be prevented by inhibition of calcium influx [69]. The increased activity of calcium-dependent kinases or altered glutamate homeostasis can enhance tau phosphorylation [70, 71], meaning the glutamate-induced excitotoxicity can increase dysfunctional tau expression. On the other hand, several other studies find that tau also plays a critical role in eliciting excitotoxicity [72–77]. There is an increase in KCL-evoked glutamate release and a decrease in glutamate clearance in TauP301L mice [74]. The molecular mechanisms underlying tau-induced excitotoxicity remain elucidated. A latter study demonstrates tau facilitates excitotoxicity with a mechanism that does not directly involve facilitation of calcium influx through kainic acid (KA) receptors [78]. However, another study suggests that the reduction of the pY18-tau formation or level can depress excitotoxicity by diminishing N-methyl-D-aspartic acid (NMDA) receptor-dependent calcium influx [79, 80]. Altogether, excitotoxicity and tau phosphorylation lead to a vicious circle in promoting cell death in ischemic brain.
Tau protein and inflammation
Inflammation of neural tissue, also called neuroinflammation, is considered the main cause of mortality in ischemia/reperfusion stroke [81]. Some previous studies have suggested that dysfunctional tau is closely related to inflammatory cascade. The inflammatory messengers can significantly affect the structure and function of tau [82–84]. The misfolded tau can represent a trigger for inflammatory cascade [82–84]. The exact roles of inflammatory processes on tau pathology or dysfunctional tau on inflammation still remain unequivocal. Some researchers generally consider inflammation an exacerbating factor [83], but another study also shows that acute inflammation may decrease the oligomeric tau levels by improving the ability of microglia [85]. The first direct evidence for the role of inflammation on tau pathology was demonstrated in a vitro study in 2003. This study showed that the inflammatory mediator, interleukin-1β (IL-1β), could promote tau phosphorylation via activating p38-mitogen-activated protein kinases (MAPK) [86]. In the same year, this role was confirmed in a vivo study with the 3xTg model [87]. The latter studies also showed that a series of bacterial or viral immune stressors and tumor necrosis factors could trigger an increase in tau phosphorylation [88–90]. So reducing tau levels or inhibiting inflammatory pathways could serve as a way to resist tauopathies [91]. In 2009, Kovac et al. found a novel toxic gain of function of misfolded tau, truncated tau. Truncated tau could induce significant decrease of trans-endothelial electrical resistance and increase of endothelial permeability of BBB. Further, researchers also found that truncated tau showed cytotoxic effects on astrocyte-microglia culture manifested by increased extracellular adenylate kinase levels. Blood-brain barrier damage induced by truncated tau was mediated through pro-inflammatory cytokine TNF-α and chemokine MCP-1 [23]. It is noteworthy that pro-inflammatory cytokine interferon-γ (IFNγ) has been reported to have opposing effects on the phosphorylation and dephosphorylation of tau [92]. The macrophages and microglia play a vital role in neuroinflammation. Tau oligomers can only be phagocytosed by both macrophages and microglia under physiological condition [85]. Microglial internalization has been indicated to be effective to both soluble and aggregated human tau [93]. Overall, suppressing the inflammation in neural tissue may prove paradoxically effective in the development of tau pathology. Further studies are required to elucidate the molecular mechanism.
Tau protein and angiogenesis
Vascular endothelium refers to cells that line the entire circulatory system. It has a close relationship with thrombosis and thrombolysis. Dysfunctional endothelium plays a key role in the pathology of stroke by increasing the atherosclerotic plaques size and vulnerability [94]. Previous studies have suggested that endothelial cells can be damaged by tauopathy, such as hyperphosphorylation and insolubility, via decreasing microtubule assembly [95–98]. Measurement of cerebral perfusion in different studies indicate that tau pathology is related to reduced blood flow [99, 100]. Truncated tau has been proven to play an important role in regulating permeability of BBB by decreasing transendothelial electrical resistance (TEER) and increasing mannitol permeability [23]. In aged tau-overexpressing mice, tau pathological changes can impact the brain endothelial cell biological function by influencing the integrity of the brain’s microvasculature [101]. Furthermore, researchers in this study also find the accumulation of pathological tau is related to the expression of hypoxia-and/or angiogenesis-related genes, such as Serpine1, Vegfa, Plau and Hmox1 [101]. However, the precise cellular signals of these changes and the specific interactions between tau and endothelial cells still remain further elucidated. Therefore, tau pathology may play an important role in the process of BBB disruption and neurogenesis by regulating activities of endothelial cells after ischemic stroke.
Tau protein and mitochondrial dysfunction
Neuronal cells are particularly sensitive to energy deficiencies. The function of mitochondria is to maintain the energy supply for cells. Mitochondrial dysfunction is one of the pivotal pathological processes in brain ischemia and reperfusion. Mitochondrial dysfunction then causes neurons necrosis, autophagy and apoptosis [102]. Disruption of mitochondrial dynamics (the balance between fission and fusion) is the core factor in mitochondrial dysfunction. Previous studies showed that dynamin-related protein 1 (DRP1), a kind of mitochondrial fission proteins, could interact with phosphorylated tau, leading to mitochondrial dysfunction [103, 104]. Meanwhile, reducing Drp1 levels could protect against mitochondrial dysfunction induced by hyperphosphorylated tau [105]. Additionally, a significant association between tau accumulation and mitochondrial translocation deficits was found both in the mouse models and human brains [106]. The abnormal mitochondrial trafficking can be improved through reducing soluble tau levels [106]. In cell and animal studies, overexpressed tau can both destroy physiological function and distribution of mitochondria, which may cause ATP exhausting, oxidative stress and synaptic dysfunction [107–109]. In the mechanism studies, glycogen synthase kinase 3 (GSK3), axonal protein phosphatase 1 (PP1), and phosphorylated tau trapped kinesin motor protein complex JIP1 were considered to be involved in the pathological interaction [110, 111]. It is interesting to notice that tau phosphorylation can also be aggravated by ROS mimicking mitochondrial oxidative stress in neuronal cells [112]. Altogether, tau pathology can destroy the mitochondrial dynamics and function, while the dysfunctional mitochondria may indicate tau phosphorylation and aggregation.
Tau protein and neurovascular unit damage
The abnormal neuron-to-neuron connections and dysfunctional interactions among the different components in the neurovascular unit (NVU) might be the main reasons for functional deficits after ischemic stroke [113]. A study found that ischemia could induce neurovascular alterations, glial changes, and the loss of tight junctions in NVU, leading to the BBB breakdown [9]. By immunofluorescence assay, they also confirmed the Aβdeposits and dysfunctional tau existed with glial reactions and morphologically altered endothelia [9]. Therefore, tau may play an important role in the process of NVU damage after ischemic stroke. In the future, a focus on all components and investigation of intercellular signaling and signaling between cells and extracellular matrix is essential to clarify all the facts about ischemic stroke.
In summary, we have discussed the potential mechanisms of tau in ischemic stroke, including oxidative stress, apoptosis, autophagy, excitotoxicity, inflammation, endothelium and angiogenesis, and mitochondrial dysfunction. In addition, we also discussed the role of tau in NVU damage. Tau may stand at the intersection of multiple regulatory mechanisms for major pathological changes in ischemic stroke.
Therapeutic researches
From the above, it is clear that intervention in tau-mediated pathological changes could be considered as a clinically beneficial strategy in ischemic stroke. No such therapy related to tau-regulation have yet achieved regulatory approval for clinical application and further evidence is still required. However, there has been many studies achieved encouraging progress. They found the reduction in tau activities and levels might have clinical benefits in stroke treatment. In this part, we will mainly discuss the findings in animal and clinical studies.
Tau in animal studies
In animal studies, tau hyperphosphorylation was found in rats after ischemic damage, and this was considered the consequence of the activation/ inactivation of a variety of phosphatases and kinases [31]. In addition, tau hyperphosphorylation could be caused by hypoxia-dependent mechanism in vascular dysfunction models, such as the ischemic model [114]. Focal mild hypothermia is considered a protective factor on ischemia/reperfusion damage. It can significantly reduce the neurotoxicity by influencing the level of tau in rats [115]. In 2017, two important preclinical studies involving transient middle cerebral artery occlusion (MCAO) mouse models suggested novel roles for tau in acute ischemic injury, indicating that agents targeting tau and related proteins have the potential to reduce the severity of acute brain damage following stroke [10, 11]. Peng Lei and colleagues found no elevated brain iron or reperfusion injury in young (3-month-old) tau–/– mice after MCAO. While this protection was lost in older (12-month-old) tau–/– mice: the brain iron accumulated rapidly. However, the protective effects of tau knockout could be revived through normalizing the iron elevation during the reperfusion phase. They suggested the interaction between tau and iron might be pleiotropic modulators of ischemic stroke [10]. Ittner and co-workers found no up-regulation of the immediate-early genes Arc, Fos and Junb in tau–/– mice after MCAO damage. But the levels of their mRNA were higher in tau+/+ brains [11]. They also demonstrated several signaling pathways were differently activated between tau–/– mice and tau+/+ mice, mitogen-activated protein kinase (MAPK) pathway was the most notable one. Then, the inhibitor of excitotoxic RAS/ERK signaling in tau–/– mice, SynGAP1, was found significantly increased at the post-synapse in the investigation of the MAPK pathway. This study demonstrated that tau and SynGAP1 might be potential targets for acute ischemic stroke [11]. Some other studies also show that inhibitor 2 of protein phosphatase 2A (I2PP2A) can produce hyperphosphorylation of tau through inhibition of PP2A [116] in MCAO mouse model [117]. Increased level of glutamate transporter 1 in transgenic mouse model can reduce ischemic brain damage through reducing the accumulation of extracellular glutamate and the activation of subsequent calpain and caspase [118].
Tau in clinical studies
In clinical studies, an increase of the total tau level was found in human cerebrospinal fluid(CSF) after brain injury, including ischemic stroke [119, 120]. Meanwhile, tau was found measurable in serum within 6 h after ischemic symptom onset [121]. The concentration might peak after 3–5 days [121], or later [122]. Moreover, there was no statistical correlation between tau serum levels and the severity of clinical deficit or disability as assessed by the Barthel index (BI). But the serum levels of tau were correlated with infarct volumes (from 7ml to 48ml) and functional outcomes after 90 days [121]. The results were consistent with other studies which indicated that the absence of tau in serum during the acute phase (<24h) of ischemia could predict good clinical outcomes in 90 days after stroke [123]. Patients with detected tau in serum got more severe neurological deficits and worse functional outcomes when compared with patients without tau [124]. However, other researchers discovered tau protein levels were correlated with the scored neurological deficits (BI) from 48 h onward. They also found the serum tau levels had no significant correlation with stroke etiology as represented by the TOAST-criteria [125]. A recent prospective study proved that tau levels were closely related to not only stroke severity as assessed by NIHSS, but also long-term outcomes both in plasma and CSF [126]. Notably, the study of autopsied brains from patients with cerebral infarction found that an increase of tau immunoreactivity and deposition of tau in ischemic area, but tau deposits were not organized into fibrils or more solid inclusions indicating that tau epitope was secondary to ischemic damage [127, 128]. Nevertheless, tau can be detected in the serum of approximately 40% of stroke patients [121, 122]. Present studies have not proven why tau could not be detected in the blood of all stroke patients. Some researchers think tau may occur in blood due to the disruption of BBB. Some factors like MMP9 may play a key role in the release of tau into circulation [122].
At present, researchers have found several methods to reduce the tau aggregation or tau levels. Methylene blue was considered a tau aggregation blocker. It could induce autophagy and attenuate tauopathy in vitro and in vivo [129], to block tau aggregation in C. elegans [130], although its exact mechanism of action is still not understood. The AMPK-related kinase Nuak1 has been identified as a regulator of tau levels. Inhibition of Nuak1 in fruit flies suppressed tau-dependent neurodegeneration [131]. Moreover, several approaches have targeted the putative enzymes that are responsible for tau changes, such as ERK inhibitor [132], JNK inhibitor [133], GSK3β [134]. However, these methods have not been used clinically for ischemic stroke. Further studies need to explore the application of tau-based therapeutic strategies, especially in acute phase of ischemic stroke.
Conclusions and perspectives
This review is committed to describe the pathological roles of tau following cerebral ischemia. Tau is a protein that plays a vital role not only in microtubule assembly and stabilization, but also in pathophysiology of ischemic stroke. Initially, we provided a general aspects of tau protein, including descriptions of its structure, physiological functions and pathological functions. Then, we introduce different pathological states of tau protein under ischemic condition. The pathological changes (such as oxidative stress, autophagy, excitotoxicity, inflammation, endothelium and angiogenesis, and mitochondrial dysfunction) of tau protein determine its potential regulatory mechanisms in ischemic stroke. Phosphorylation is the main pathological change of tau in ischemic stroke. Therefore, controlling tau phosphorylation may induce more protective effects under ischemic stimuli. As some experimental results are from mouse model with FTDP-17 mutations, there might be differences between mouse model with FTDP-17 mutations and those with ischemic injury in pathogenetic mechanisms leading to degeneration. Some studies proved that the regional redistribution of tau from the neuropil to neuronal perikarya in their stroke model was thought to share similarity with that occurring in Alzheimer's disease [30]. But the results of molecular changes in FTDP-17 mutations mouse might different in mouse with stroke. Therefore, more researches still need to explore molecular mechanisms in mouse with ischemic injury. Lastly, we discuss about the therapeutic researches on the treatment of stroke with tau protein. The animal studies indicate a role for tau protein in acute ischemic brain damage, suggesting that agents targeting tau and related proteins have the great potential to reduce the severity of brain damage following acute ischemic stroke. The clinical studies show that the level of serum/plasma or CSF tau is related to the stroke severity of clinical deficit and long-term outcomes. The underlying mechanisms of pathological tau-induced side effects during and after ischemia/reperfusion process are complex. There are insufficient clinical studies focused on link between tau protein and ischemic stroke. However, we still believe that revealing the molecular mechanisms of tau in cerebral ischemia and regulating the tau phosphorylation may be conductive to developing a potential novel target for the ischemic stroke therapy.
Acknowledgments
We thank Dr. Zhang for helping with the preparation of the Figures in this manuscript.
Conflicts of Interest
The authors declare that they have no competing interests.
Funding
This work was supported by Important Weak Subject Construction Project of Pudong Health and Family Planning Commission of Shanghai (Grant No. PWZbr2017-06).
References
- 1. Wang H, Naghavi M, Allen C, Barber RM, Bhutta ZA, Carter A, Casey DC, Charlson FJ, Chen AZ, Coates MM, Coggeshall M, Dandona L, Dicker DJ, et al, and GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016; 388:1459–544. https://doi.org/10.1016/S0140-6736(16)31012-1 [PubMed]
- 2. Shobha N, Buchan AM, Hill MD, and Canadian Alteplase for Stroke Effectiveness Study (CASES). Thrombolysis at 3-4.5 hours after acute ischemic stroke onset--evidence from the Canadian Alteplase for Stroke Effectiveness Study (CASES) registry. Cerebrovasc Dis. 2011; 31:223–28. https://doi.org/10.1159/000321893 [PubMed]
- 3. De Meyer SF, Stoll G, Wagner DD, Kleinschnitz C. von Willebrand factor: an emerging target in stroke therapy. Stroke. 2012; 43:599–606. https://doi.org/10.1161/STROKEAHA.111.628867 [PubMed]
- 4. Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989; 8:393–99. https://doi.org/10.1002/j.1460-2075.1989.tb03390.x [PubMed]
- 5. Gao YL, Wang N, Sun FR, Cao XP, Zhang W, Yu JT. Tau in neurodegenerative disease. Ann Transl Med. 2018; 6:175. https://doi.org/10.21037/atm.2018.04.23 [PubMed]
- 6. Goedert M, Eisenberg DS, Crowther RA. Propagation of Tau Aggregates and Neurodegeneration. Annu Rev Neurosci. 2017; 40:189–210. https://doi.org/10.1146/annurev-neuro-072116-031153 [PubMed]
- 7. Goedert M, Jakes R, Spillantini MG. The Synucleinopathies: Twenty Years On. J Parkinsons Dis. 2017; 7:S51–69. https://doi.org/10.3233/JPD-179005 [PubMed]
- 8. Gordon-Krajcer W, Kozniewska E, Lazarewicz JW, Ksiezak-Reding H. Differential changes in phosphorylation of tau at PHF-1 and 12E8 epitopes during brain ischemia and reperfusion in gerbils. Neurochem Res. 2007; 32:729–37. https://doi.org/10.1007/s11064-006-9199-3 [PubMed]
- 9. Michalski D, Hofmann S, Pitsch R, Grosche J, Härtig W. Neurovascular Specifications in the Alzheimer-Like Brain of Mice Affected by Focal Cerebral Ischemia: Implications for Future Therapies. J Alzheimers Dis. 2017; 59:655–74. https://doi.org/10.3233/JAD-170185 [PubMed]
- 10. Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL, Liuyang ZY, Roisman L, Zhang ST, Ayton S, Wang Q, Crouch PJ, Ganio K, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017; 22:1520–30. https://doi.org/10.1038/mp.2017.171 [PubMed]
- 11. Bi M, Gladbach A, van Eersel J, Ittner A, Przybyla M, van Hummel A, Chua SW, van der Hoven J, Lee WS, Müller J, Parmar J, Jonquieres GV, Stefen H, et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun. 2017; 8:473. https://doi.org/10.1038/s41467-017-00618-0 [PubMed]
- 12. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975; 72:1858–62. https://doi.org/10.1073/pnas.72.5.1858 [PubMed]
- 13. Trojanowski JQ, Schuck T, Schmidt ML, Lee VM. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem. 1989; 37:209–15. https://doi.org/10.1177/37.2.2492045 [PubMed]
- 14. Tashiro K, Hasegawa M, Ihara Y, Iwatsubo T. Somatodendritic localization of phosphorylated tau in neonatal and adult rat cerebral cortex. Neuroreport. 1997; 8:2797–801. https://doi.org/10.1097/00001756-199708180-00029 [PubMed]
- 15. Gu Y, Oyama F, Ihara Y. Tau is widely expressed in rat tissues. J Neurochem. 1996; 67:1235–44. https://doi.org/10.1046/j.1471-4159.1996.67031235.x [PubMed]
- 16. Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011; 70:410–26. https://doi.org/10.1016/j.neuron.2011.04.009 [PubMed]
- 17. Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001; 24:1121–59. https://doi.org/10.1146/annurev.neuro.24.1.1121 [PubMed]
- 18. Hong XP, Peng CX, Wei W, Tian Q, Liu YH, Yao XQ, Zhang Y, Cao FY, Wang Q, Wang JZ. Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus. 2010; 20:1339–49. https://doi.org/10.1002/hipo.20712 [PubMed]
- 19. Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008; 319:1086–89. https://doi.org/10.1126/science.1152993 [PubMed]
- 20. Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, Talahari S, Nesslany F, Lefebvre B, Bonnefoy E, Buée L, Galas MC. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci. 2014; 8:84. https://doi.org/10.3389/fncel.2014.00084 [PubMed]
- 21. Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, Finucane MM, Sanchez PE, Mucke L. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurol. 2014; 76:443–56. https://doi.org/10.1002/ana.24230 [PubMed]
- 22. Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem. 1996; 271:28741–44. https://doi.org/10.1074/jbc.271.46.28741 [PubMed]
- 23. Kovac A, Zilkova M, Deli MA, Zilka N, Novak M. Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J Alzheimers Dis. 2009; 18:897–906. https://doi.org/10.3233/JAD-2009-1197 [PubMed]
- 24. Pluta R, Jabłoński M, Ułamek-Kozioł M, Kocki J, Brzozowska J, Januszewski S, Furmaga-Jabłońska W, Bogucka-Kocka A, Maciejewski R, Czuczwar SJ. Sporadic Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol Neurobiol. 2013; 48:500–15. https://doi.org/10.1007/s12035-013-8439-1 [PubMed]
- 25. Pluta R, Bogucka-Kocka A, Ułamek-Kozioł M, Bogucki J, Januszewski S, Kocki J, Czuczwar SJ. Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer’s phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer’s disease. Pharmacol Rep. 2018; 70:881–84. https://doi.org/10.1016/j.pharep.2018.03.004 [PubMed]
- 26. Kato T, Hirano A, Katagiri T, Sasaki H, Yamada S. Neurofibrillary tangle formation in the nucleus basalis of Meynert ipsilateral to a massive cerebral infarct. Ann Neurol. 1988; 23:620–23. https://doi.org/10.1002/ana.410230617 [PubMed]
- 27. Khan S, Yuldasheva NY, Batten TF, Pickles AR, Kellett KA, Saha S. Tau pathology and neurochemical changes associated with memory dysfunction in an optimised murine model of global cerebral ischaemia - A potential model for vascular dementia? Neurochem Int. 2018; 118:134–44. https://doi.org/10.1016/j.neuint.2018.04.004 [PubMed]
- 28. Whitehead SN, Hachinski VC, Cechetto DF. Interaction between a rat model of cerebral ischemia and beta-amyloid toxicity: inflammatory responses. Stroke. 2005; 36:107–12. https://doi.org/10.1161/01.STR.0000149627.30763.f9 [PubMed]
- 29. Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. 2007; 1772:473–83. https://doi.org/10.1016/j.bbadis.2006.10.011 [PubMed]
- 30. Fujii H, Takahashi T, Mukai T, Tanaka S, Hosomi N, Maruyama H, Sakai N, Matsumoto M. Modifications of tau protein after cerebral ischemia and reperfusion in rats are similar to those occurring in Alzheimer’s disease - Hyperphosphorylation and cleavage of 4- and 3-repeat tau. J Cereb Blood Flow Metab. 2017; 37:2441–57. https://doi.org/10.1177/0271678X16668889 [PubMed]
- 31. Wen Y, Yang S, Liu R, Simpkins JW. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004; 1022:30–38. https://doi.org/10.1016/j.brainres.2004.05.106 [PubMed]
- 32. Morioka M, Kawano T, Yano S, Kai Y, Tsuiki H, Yoshinaga Y, Matsumoto J, Maeda T, Hamada J, Yamamoto H, Fukunaga K, Kuratsu J. Hyperphosphorylation at serine 199/202 of tau factor in the gerbil hippocampus after transient forebrain ischemia. Biochem Biophys Res Commun. 2006; 347:273–78. https://doi.org/10.1016/j.bbrc.2006.06.096 [PubMed]
- 33. Wen Y, Yang S, Liu R, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer’s disease-like tauopathy in female rats. J Biol Chem. 2004; 279:22684–92. https://doi.org/10.1074/jbc.M311768200 [PubMed]
- 34. Kovalska M, Tothova B, Kovalska L, Tatarkova Z, Kalenska D, Tomascova A, Adamkov M, Lehotsky J. Association of Induced Hyperhomocysteinemia with Alzheimer’s Disease-Like Neurodegeneration in Rat Cortical Neurons After Global Ischemia-Reperfusion Injury. Neurochem Res. 2018; 43:1766–78. https://doi.org/10.1007/s11064-018-2592-x [PubMed]
- 35. Burkhart KK, Beard DC, Lehman RA, Billingsley ML. Alterations in tau phosphorylation in rat and human neocortical brain slices following hypoxia and glucose deprivation. Exp Neurol. 1998; 154:464–72. https://doi.org/10.1006/exnr.1998.6899 [PubMed]
- 36. Dewar D, Dawson D. Tau protein is altered by focal cerebral ischaemia in the rat: an immunohistochemical and immunoblotting study. Brain Res. 1995; 684:70–78. https://doi.org/10.1016/0006-8993(95)00417-O [PubMed]
- 37. Geddes JW, Schwab C, Craddock S, Wilson JL, Pettigrew LC. Alterations in tau immunostaining in the rat hippocampus following transient cerebral ischemia. J Cereb Blood Flow Metab. 1994; 14:554–64. https://doi.org/10.1038/jcbfm.1994.69 [PubMed]
- 38. Majd S, Power JH, Koblar SA, Grantham HJ. Early glycogen synthase kinase-3β and protein phosphatase 2A independent tau dephosphorylation during global brain ischaemia and reperfusion following cardiac arrest and the role of the adenosine monophosphate kinase pathway. Eur J Neurosci. 2016; 44:1987–97. https://doi.org/10.1111/ejn.13277 [PubMed]
- 39. Shackelford DA, Yeh RY. Dephosphorylation of tau during transient forebrain ischemia in the rat. Mol Chem Neuropathol. 1998; 34:103–20. https://doi.org/10.1007/BF02815073 [PubMed]
- 40. Mailliot C, Podevin-Dimster V, Rosenthal RE, Sergeant N, Delacourte A, Fiskum G, Buée L. Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2000; 20:543–49. https://doi.org/10.1097/00004647-200003000-00013 [PubMed]
- 41. Goedert M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018; 592:2383–91. https://doi.org/10.1002/1873-3468.13108 [PubMed]
- 42. Chen S, Liu AR, An FM, Yao WB, Gao XD. Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer’s disease by exendin-4. Age (Dordr). 2012; 34:1211–24. https://doi.org/10.1007/s11357-011-9303-8 [PubMed]
- 43. Zhou S, Yu G, Chi L, Zhu J, Zhang W, Zhang Y, Zhang L. Neuroprotective effects of edaravone on cognitive deficit, oxidative stress and tau hyperphosphorylation induced by intracerebroventricular streptozotocin in rats. Neurotoxicology. 2013; 38:136–45. https://doi.org/10.1016/j.neuro.2013.07.007 [PubMed]
- 44. Correia SC, Santos RX, Santos MS, Casadesus G, Lamanna JC, Perry G, Smith MA, Moreira PI. Mitochondrial abnormalities in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. Curr Alzheimer Res. 2013; 10:406–19. https://doi.org/10.2174/1567205011310040006 [PubMed]
- 45. Kang SW, Kim SJ, Kim MS. Oxidative stress with tau hyperphosphorylation in memory impaired 1,2-diacetylbenzene-treated mice. Toxicol Lett. 2017; 279:53–59. https://doi.org/10.1016/j.toxlet.2017.07.892 [PubMed]
- 46. Clausen A, Xu X, Bi X, Baudry M. Effects of the superoxide dismutase/catalase mimetic EUK-207 in a mouse model of Alzheimer’s disease: protection against and interruption of progression of amyloid and tau pathology and cognitive decline. J Alzheimers Dis. 2012; 30:183–208. https://doi.org/10.3233/JAD-2012-111298 [PubMed]
- 47. Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, Schilling B, Mavros C, Masters CL, Volitakis I, Li QX, Laughton K, Hubbard A, et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One. 2007; 2:e536. https://doi.org/10.1371/journal.pone.0000536 [PubMed]
- 48. Alavi Naini SM, Soussi-Yanicostas N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid Med Cell Longev. 2015; 2015:151979. https://doi.org/10.1155/2015/151979 [PubMed]
- 49. Gusev GP, Govekar R, Gadewal N, Agalakova NI. Understanding quasi-apoptosis of the most numerous enucleated components of blood needs detailed molecular autopsy. Ageing Res Rev. 2017; 35:46–62. https://doi.org/10.1016/j.arr.2017.01.002 [PubMed]
- 50. Cheng W, Chen W, Wang P, Chu J. Asiatic acid protects differentiated PC12 cells from Aβ25-35-induced apoptosis and tau hyperphosphorylation via regulating PI3K/Akt/GSK-3β signaling. Life Sci. 2018; 208:96–101. https://doi.org/10.1016/j.lfs.2018.07.016 [PubMed]
- 51. Ma X, Liu L, Meng J. MicroRNA-125b promotes neurons cell apoptosis and Tau phosphorylation in Alzheimer’s disease. Neurosci Lett. 2017; 661:57–62. https://doi.org/10.1016/j.neulet.2017.09.043 [PubMed]
- 52. Xiao N, Zhang F, Zhu B, Liu C, Lin Z, Wang H, Xie WB. CDK5-mediated tau accumulation triggers methamphetamine-induced neuronal apoptosis via endoplasmic reticulum-associated degradation pathway. Toxicol Lett. 2018; 292:97–107. https://doi.org/10.1016/j.toxlet.2018.04.027 [PubMed]
- 53. Maday S, Holzbaur EL. Compartment-Specific Regulation of Autophagy in Primary Neurons. J Neurosci. 2016; 36:5933–45. https://doi.org/10.1523/JNEUROSCI.4401-15.2016 [PubMed]
- 54. Vidal RL, Matus S, Bargsted L, Hetz C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol Sci. 2014; 35:583–91. https://doi.org/10.1016/j.tips.2014.09.002 [PubMed]
- 55. Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008; 28:6926–37. https://doi.org/10.1523/JNEUROSCI.0800-08.2008 [PubMed]
- 56. Wu H, Niu H, Wu C, Li Y, Wang K, Zhang J, Wang Y, Yang S. The autophagy-lysosomal system in subarachnoid haemorrhage. J Cell Mol Med. 2016; 20:1770–78. https://doi.org/10.1111/jcmm.12855 [PubMed]
- 57. Feng J, Chen X, Shen J. Reactive nitrogen species as therapeutic targets for autophagy: implication for ischemic stroke. Expert Opin Ther Targets. 2017; 21:305–17. https://doi.org/10.1080/14728222.2017.1281250 [PubMed]
- 58. Koike MA, Green KN, Blurton-Jones M, Laferla FM. Oligemic hypoperfusion differentially affects tau and amyloid-beta. Am J Pathol. 2010; 177:300–10. https://doi.org/10.2353/ajpath.2010.090750 [PubMed]
- 59. Huuskonen MT, Loppi S, Dhungana H, Keksa-Goldsteine V, Lemarchant S, Korhonen P, Wojciechowski S, Pollari E, Valonen P, Koponen J, Takashima A, Landreth G, Goldsteins G, et al. Bexarotene targets autophagy and is protective against thromboembolic stroke in aged mice with tauopathy. Sci Rep. 2016; 6:33176. https://doi.org/10.1038/srep33176 [PubMed]
- 60. Falcon B, Noad J, McMahon H, Randow F, Goedert M. Galectin-8-mediated selective autophagy protects against seeded tau aggregation. J Biol Chem. 2018; 293:2438–51. https://doi.org/10.1074/jbc.M117.809293 [PubMed]
- 61. Scott IS, Lowe JS. The ubiquitin-binding protein p62 identifies argyrophilic grain pathology with greater sensitivity than conventional silver stains. Acta Neuropathol. 2007; 113:417–20. https://doi.org/10.1007/s00401-006-0165-6 [PubMed]
- 62. Ozcelik S, Fraser G, Castets P, Schaeffer V, Skachokova Z, Breu K, Clavaguera F, Sinnreich M, Kappos L, Goedert M, Tolnay M, Winkler DT. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One. 2013; 8:e62459. https://doi.org/10.1371/journal.pone.0062459 [PubMed]
- 63. Xu Y, Martini-Stoica H, Zheng H. A seeding based cellular assay of tauopathy. Mol Neurodegener. 2016; 11:32. https://doi.org/10.1186/s13024-016-0100-9 [PubMed]
- 64. Hasegawa M. Molecular Mechanisms in the Pathogenesis of Alzheimer’s disease and Tauopathies-Prion-Like Seeded Aggregation and Phosphorylation. Biomolecules. 2016; 6:E24. https://doi.org/10.3390/biom6020024 [PubMed]
- 65. Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015; 41:24–46. https://doi.org/10.1111/nan.12213 [PubMed]
- 66. Ojo OB, Amoo ZA, Saliu IO, Olaleye MT, Farombi EO, Akinmoladun AC. Neurotherapeutic potential of kolaviron on neurotransmitter dysregulation, excitotoxicity, mitochondrial electron transport chain dysfunction and redox imbalance in 2-VO brain ischemia/reperfusion injury. Biomed Pharmacother. 2019; 111:859–72. https://doi.org/10.1016/j.biopha.2018.12.144 [PubMed]
- 67. Yin A, Guo H, Tao L, Cai G, Wang Y, Yao L, Xiong L, Zhang J, Li Y. NDRG2 Protects the Brain from Excitotoxicity by Facilitating Interstitial Glutamate Uptake. Transl Stroke Res. 2019. [Epub ahead of print]. https://doi.org/10.1007/s12975-019-00708-9 [PubMed]
- 68. Tejeda GS, Esteban-Ortega GM, San Antonio E, Vidaurre OG, Díaz-Guerra M. Prevention of excitotoxicity-induced processing of BDNF receptor TrkB-FL leads to stroke neuroprotection. EMBO Mol Med. 2019; 11:e9950. https://doi.org/10.15252/emmm.201809950 [PubMed]
- 69. Ho PI, Ortiz D, Rogers E, Shea TB. Multiple aspects of homocysteine neurotoxicity: glutamate excitotoxicity, kinase hyperactivation and DNA damage. J Neurosci Res. 2002; 70:694–702. https://doi.org/10.1002/jnr.10416 [PubMed]
- 70. Ekinci FJ, Malik KU, Shea TB. Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to beta-amyloid. MAP kinase mediates beta-amyloid-induced neurodegeneration. J Biol Chem. 1999; 274:30322–27. https://doi.org/10.1074/jbc.274.42.30322 [PubMed]
- 71. Petroni D, Tsai J, Mondal D, George W. Attenuation of low dose methylmercury and glutamate induced-cytotoxicity and tau phosphorylation by an N-methyl-D-aspartate antagonist in human neuroblastoma (SHSY5Y) cells. Environ Toxicol. 2013; 28:700–06. https://doi.org/10.1002/tox.20765 [PubMed]
- 72. Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, Noebels JL. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci. 2013; 33:1651–59. https://doi.org/10.1523/JNEUROSCI.3191-12.2013 [PubMed]
- 73. DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, Bennett CF, Cirrito JR, Holtzman DM, Miller TM. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013; 33:12887–97. https://doi.org/10.1523/JNEUROSCI.2107-13.2013 [PubMed]
- 74. Hunsberger HC, Rudy CC, Batten SR, Gerhardt GA, Reed MN. P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J Neurochem. 2015; 132:169–82. https://doi.org/10.1111/jnc.12967 [PubMed]
- 75. Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol. 2013; 698:6–18. https://doi.org/10.1016/j.ejphar.2012.10.032 [PubMed]
- 76. Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010; 11:682–96. https://doi.org/10.1038/nrn2911 [PubMed]
- 77. Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci USA. 2006; 103:2892–97. https://doi.org/10.1073/pnas.0511065103 [PubMed]
- 78. Pallo SP, DiMaio J, Cook A, Nilsson B, Johnson GV. Mechanisms of tau and Aβ-induced excitotoxicity. Brain Res. 2016; 1634:119–31. https://doi.org/10.1016/j.brainres.2015.12.048 [PubMed]
- 79. Miyamoto T, Stein L, Thomas R, Djukic B, Taneja P, Knox J, Vossel K, Mucke L. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol Neurodegener. 2017; 12:41. https://doi.org/10.1186/s13024-017-0176-x [PubMed]
- 80. Decker JM, Krüger L, Sydow A, Dennissen FJ, Siskova Z, Mandelkow E, Mandelkow EM. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 2016; 17:552–69. https://doi.org/10.15252/embr.201541439 [PubMed]
- 81. Chen YJ, Nguyen HM, Maezawa I, Grössinger EM, Garing AL, Köhler R, Jin LW, Wulff H. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J Cereb Blood Flow Metab. 2016; 36:2146–61. https://doi.org/10.1177/0271678X15611434 [PubMed]
- 82. Kovac A, Zilka N, Kazmerova Z, Cente M, Zilkova M, Novak M. Misfolded truncated protein τ induces innate immune response via MAPK pathway. J Immunol. 2011; 187:2732–39. https://doi.org/10.4049/jimmunol.1100216 [PubMed]
- 83. Zilka N, Kazmerova Z, Jadhav S, Neradil P, Madari A, Obetkova D, Bugos O, Novak M. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012; 9:47. https://doi.org/10.1186/1742-2094-9-47 [PubMed]
- 84. Asai H, Ikezu S, Woodbury ME, Yonemoto GM, Cui L, Ikezu T. Accelerated neurodegeneration and neuroinflammation in transgenic mice expressing P301L tau mutant and tau-tubulin kinase 1. Am J Pathol. 2014; 184:808–18. https://doi.org/10.1016/j.ajpath.2013.11.026 [PubMed]
- 85. Majerova P, Zilkova M, Kazmerova Z, Kovac A, Paholikova K, Kovacech B, Zilka N, Novak M. Microglia display modest phagocytic capacity for extracellular tau oligomers. J Neuroinflammation. 2014; 11:161. https://doi.org/10.1186/s12974-014-0161-z [PubMed]
- 86. Li Y, Liu L, Barger SW, Griffin WS. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003; 23:1605–11. https://doi.org/10.1523/JNEUROSCI.23-05-01605.2003 [PubMed]
- 87. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003; 39:409–21. https://doi.org/10.1016/S0896-6273(03)00434-3 [PubMed]
- 88. Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005; 25:8843–53. https://doi.org/10.1523/JNEUROSCI.2868-05.2005 [PubMed]
- 89. Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011; 178:2811–22. https://doi.org/10.1016/j.ajpath.2011.02.012 [PubMed]
- 90. Janelsins MC, Mastrangelo MA, Park KM, Sudol KL, Narrow WC, Oddo S, LaFerla FM, Callahan LM, Federoff HJ, Bowers WJ. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol. 2008; 173:1768–82. https://doi.org/10.2353/ajpath.2008.080528 [PubMed]
- 91. Maphis N, Xu G, Kokiko-Cochran ON, Cardona AE, Ransohoff RM, Lamb BT, Bhaskar K. Loss of tau rescues inflammation-mediated neurodegeneration. Front Neurosci. 2015; 9:196. https://doi.org/10.3389/fnins.2015.00196 [PubMed]
- 92. Mastrangelo MA, Sudol KL, Narrow WC, Bowers WJ. Interferon-gamma differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am J Pathol. 2009; 175:2076–88. https://doi.org/10.2353/ajpath.2009.090059 [PubMed]
- 93. Bolós M, Llorens-Martín M, Jurado-Arjona J, Hernández F, Rábano A, Avila J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J Alzheimers Dis. 2016; 50:77–87. https://doi.org/10.3233/JAD-150704 [PubMed]
- 94. Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, Nishigaki I. The vascular endothelium and human diseases. Int J Biol Sci. 2013; 9:1057–69. https://doi.org/10.7150/ijbs.7502 [PubMed]
- 95. Göttle M, Dove S, Kees F, Schlossmann J, Geduhn J, König B, Shen Y, Tang WJ, Kaever V, Seifert R. Cytidylyl and uridylyl cyclase activity of bacillus anthracis edema factor and Bordetella pertussis CyaA. Biochemistry. 2010; 49:5494–503. https://doi.org/10.1021/bi100684g [PubMed]
- 96. Guo Q, Shen Y, Lee YS, Gibbs CS, Mrksich M, Tang WJ. Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 2005; 24:3190–201. https://doi.org/10.1038/sj.emboj.7600800 [PubMed]
- 97. Tang WJ, Guo Q. The adenylyl cyclase activity of anthrax edema factor. Mol Aspects Med. 2009; 30:423–30. https://doi.org/10.1016/j.mam.2009.06.001 [PubMed]
- 98. Balczon R, Prasain N, Ochoa C, Prater J, Zhu B, Alexeyev M, Sayner S, Frank DW, Stevens T. Pseudomonas aeruginosa exotoxin Y-mediated tau hyperphosphorylation impairs microtubule assembly in pulmonary microvascular endothelial cells. PLoS One. 2013; 8:e74343. https://doi.org/10.1371/journal.pone.0074343 [PubMed]
- 99. Smith R, Schöll M, Honer M, Nilsson CF, Englund E, Hansson O. Tau neuropathology correlates with FDG-PET, but not AV-1451-PET, in progressive supranuclear palsy. Acta Neuropathol. 2017; 133:149–51. https://doi.org/10.1007/s00401-016-1650-1 [PubMed]
- 100. Bradley KM, O’Sullivan VT, Soper ND, Nagy Z, King EM, Smith AD, Shepstone BJ. Cerebral perfusion SPET correlated with Braak pathological stage in Alzheimer’s disease. Brain. 2002; 125:1772–81. https://doi.org/10.1093/brain/awf185 [PubMed]
- 101. Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, Das S, Hyman BT. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc Natl Acad Sci USA. 2018; 115:E1289–98. https://doi.org/10.1073/pnas.1710329115 [PubMed]
- 102. Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013; 47:9–23. https://doi.org/10.1007/s12035-012-8344-z [PubMed]
- 103. Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, Cullen PJ, Liu J, Zhu X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med. 2016; 22:54–63. https://doi.org/10.1038/nm.3983 [PubMed]
- 104. DuBoff B, Götz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012; 75:618–32. https://doi.org/10.1016/j.neuron.2012.06.026 [PubMed]
- 105. Kandimalla R, Manczak M, Fry D, Suneetha Y, Sesaki H, Reddy PH. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet. 2016; 25:4881–97. https://doi.org/10.1093/hmg/ddw312 [PubMed]
- 106. Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP, Hyman BT, Spires-Jones TL. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am J Pathol. 2011; 179:2071–82. https://doi.org/10.1016/j.ajpath.2011.07.004 [PubMed]
- 107. Li XC, Hu Y, Wang ZH, Luo Y, Zhang Y, Liu XP, Feng Q, Wang Q, Ye K, Liu GP, Wang JZ. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep. 2016; 6:24756. https://doi.org/10.1038/srep24756 [PubMed]
- 108. Wang ZX, Tan L, Yu JT. Axonal transport defects in Alzheimer’s disease. Mol Neurobiol. 2015; 51:1309–21. https://doi.org/10.1007/s12035-014-8810-x [PubMed]
- 109. Chen Z, Zhong C. Oxidative stress in Alzheimer’s disease. Neurosci Bull. 2014; 30:271–81. https://doi.org/10.1007/s12264-013-1423-y [PubMed]
- 110. Ittner LM, Ke YD, Götz J. Phosphorylated Tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J Biol Chem. 2009; 284:20909–16. https://doi.org/10.1074/jbc.M109.014472 [PubMed]
- 111. Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y, Andreadis A, Fu Y, Brady ST, Binder LI. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci. 2011; 31:9858–68. https://doi.org/10.1523/JNEUROSCI.0560-11.2011 [PubMed]
- 112. Ibáñez-Salazar A, Bañuelos-Hernández B, Rodríguez-Leyva I, Chi-Ahumada E, Monreal-Escalante E, Jiménez-Capdeville ME, Rosales-Mendoza S. Oxidative Stress Modifies the Levels and Phosphorylation State of Tau Protein in Human Fibroblasts. Front Neurosci. 2017; 11:495. https://doi.org/10.3389/fnins.2017.00495 [PubMed]
- 113. Lo EH, Rosenberg GA. The neurovascular unit in health and disease: introduction. Stroke. 2009 (Suppl); 40:S2–3. https://doi.org/10.1161/STROKEAHA.108.534404 [PubMed]
- 114. Raz L, Bhaskar K, Weaver J, Marini S, Zhang Q, Thompson JF, Espinoza C, Iqbal S, Maphis NM, Weston L, Sillerud LO, Caprihan A, Pesko JC, et al. Hypoxia promotes tau hyperphosphorylation with associated neuropathology in vascular dysfunction. Neurobiol Dis. 2019; 126:124–36. https://doi.org/10.1016/j.nbd.2018.07.009 [PubMed]
- 115. Zhao JK, Guan FL, Duan SR, Zhao JW, Sun RH, Zhang LM, Wang DS. Effect of focal mild hypothermia on expression of MMP-9, TIMP-1, Tau-1 and β-APP in rats with cerebral ischaemia/reperfusion injury. Brain Inj. 2013; 27:1190–98. https://doi.org/10.3109/02699052.2013.804206 [PubMed]
- 116. Basurto-Islas G, Grundke-Iqbal I, Tung YC, Liu F, Iqbal K. Activation of asparaginyl endopeptidase leads to Tau hyperphosphorylation in Alzheimer disease. J Biol Chem. 2013; 288:17495–507. https://doi.org/10.1074/jbc.M112.446070 [PubMed]
- 117. Basurto-Islas G, Gu JH, Tung YC, Liu F, Iqbal K. Mechanism of Tau Hyperphosphorylation Involving Lysosomal Enzyme Asparagine Endopeptidase in a Mouse Model of Brain Ischemia. J Alzheimers Dis. 2018; 63:821–33. https://doi.org/10.3233/JAD-170715 [PubMed]
- 118. Liao G, Zhou M, Cheung S, Galeano J, Nguyen N, Baudry M, Bi X. Reduced early hypoxic/ischemic brain damage is associated with increased GLT-1 levels in mice expressing mutant (P301L) human tau. Brain Res. 2009; 1247:159–70. https://doi.org/10.1016/j.brainres.2008.10.022 [PubMed]
- 119. Hesse C, Rosengren L, Andreasen N, Davidsson P, Vanderstichele H, Vanmechelen E, Blennow K. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci Lett. 2001; 297:187–90. https://doi.org/10.1016/S0304-3940(00)01697-9 [PubMed]
- 120. Shiiya N, Kunihara T, Miyatake T, Matsuzaki K, Yasuda K. Tau protein in the cerebrospinal fluid is a marker of brain injury after aortic surgery. Ann Thorac Surg. 2004; 77:2034–38. https://doi.org/10.1016/j.athoracsur.2003.12.057 [PubMed]
- 121. Bitsch A, Horn C, Kemmling Y, Seipelt M, Hellenbrand U, Stiefel M, Ciesielczyk B, Cepek L, Bahn E, Ratzka P, Prange H, Otto M. Serum tau protein level as a marker of axonal damage in acute ischemic stroke. Eur Neurol. 2002; 47:45–51. https://doi.org/10.1159/000047946 [PubMed]
- 122. Kurzepa J, Bielewicz J, Grabarska A, Stelmasiak Z, Stryjecka-Zimmer M, Bartosik-Psujek H. Matrix metalloproteinase-9 contributes to the increase of tau protein in serum during acute ischemic stroke. J Clin Neurosci. 2010; 17:997–99. https://doi.org/10.1016/j.jocn.2010.01.005 [PubMed]
- 123. Lasek-Bal A, Jedrzejowska-Szypulka H, Rozycka J, Bal W, Kowalczyk A, Holecki M, Dulawa J, Lewin-Kowalik J. The presence of Tau protein in blood as a potential prognostic factor in stroke patients. J Physiol Pharmacol. 2016; 67:691–96. [PubMed]
- 124. Bielewicz J, Kurzepa J, Czekajska-Chehab E, Stelmasiak Z, Bartosik-Psujek H. Does serum Tau protein predict the outcome of patients with ischemic stroke? J Mol Neurosci. 2011; 43:241–45. https://doi.org/10.1007/s12031-010-9403-4 [PubMed]
- 125. Wunderlich MT, Lins H, Skalej M, Wallesch CW, Goertler M. Neuron-specific enolase and tau protein as neurobiochemical markers of neuronal damage are related to early clinical course and long-term outcome in acute ischemic stroke. Clin Neurol Neurosurg. 2006; 108:558–63. https://doi.org/10.1016/j.clineuro.2005.12.006 [PubMed]
- 126. De Vos A, Bjerke M, Brouns R, De Roeck N, Jacobs D, Van den Abbeele L, Guldolf K, Zetterberg H, Blennow K, Engelborghs S, Vanmechelen E. Neurogranin and tau in cerebrospinal fluid and plasma of patients with acute ischemic stroke. BMC Neurol. 2017; 17:170. https://doi.org/10.1186/s12883-017-0945-8 [PubMed]
- 127. Uchihara T, Nakamura A, Arai T, Ikeda K, Tsuchiya K. Microglial tau undergoes phosphorylation-independent modification after ischemia. Glia. 2004; 45:180–87. https://doi.org/10.1002/glia.10318 [PubMed]
- 128. Irving EA, Nicoll J, Graham DI, Dewar D. Increased tau immunoreactivity in oligodendrocytes following human stroke and head injury. Neurosci Lett. 1996; 213:189–92. https://doi.org/10.1016/0304-3940(96)12856-1 [PubMed]
- 129. Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, Duff KE. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012; 8:609–22. https://doi.org/10.4161/auto.19048 [PubMed]
- 130. Fatouros C, Pir GJ, Biernat J, Koushika SP, Mandelkow E, Mandelkow EM, Schmidt E, Baumeister R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum Mol Genet. 2012; 21:3587–603. https://doi.org/10.1093/hmg/dds190 [PubMed]
- 131. Lasagna-Reeves CA, de Haro M, Hao S, Park J, Rousseaux MW, Al-Ramahi I, Jafar-Nejad P, Vilanova-Velez L, See L, De Maio A, Nitschke L, Wu Z, Troncoso JC, et al. Reduction of Nuak1 Decreases Tau and Reverses Phenotypes in a Tauopathy Mouse Model. Neuron. 2016; 92:407–18. https://doi.org/10.1016/j.neuron.2016.09.022 [PubMed]
- 132. Le Corre S, Klafki HW, Plesnila N, Hübinger G, Obermeier A, Sahagún H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci USA. 2006; 103:9673–78. https://doi.org/10.1073/pnas.0602913103 [PubMed]
- 133. Tran HT, Sanchez L, Brody DL. Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J Neuropathol Exp Neurol. 2012; 71:116–29. https://doi.org/10.1097/NEN.0b013e3182456aed [PubMed]
- 134. Zhang X, Hernandez I, Rei D, Mair W, Laha JK, Cornwell ME, Cuny GD, Tsai LH, Steen JA, Kosik KS. Diaminothiazoles modify Tau phosphorylation and improve the tauopathy in mouse models. J Biol Chem. 2013; 288:22042–56. https://doi.org/10.1074/jbc.M112.436402 [PubMed]