



COVID-19 vulnerability: age, diseases, gender
COVID-19 is caused by coronavirus SARS-CoV-2. Most cases of COVID-19 are asymptomatic, but some are severe and lethal. Mortality is the simplest marker of COVID-19 vulnerability. COVID-19 vulnerability can be defined as a chance of death from COVID-19, once infected.
Age:
In all studies conducted in all countries, the mortality rate from COVID-19 increases exponentially with age [1–11]. Exact mortality rates varied in hundreds of studies because they depend on testing and therapeutic interventions. But the rule is clear: the mortality rate is increasing exponentially with age.
Age-related diseases:
Mortality is especially high in patients with pre-existing conditions [6, 9, 10, 12–23].
In Italy, 99% of patients, who died, had at least one illness https://www.bloomberg.com/news/articles/2020-03-18/99-of-those-who-died-from-virus-had-other-illness-italy-says.
In other words, infected people without pre-existing diseases do not die. This may seem paradoxical because we just discussed that age is sufficient to increase mortality exponentially. This is because pre-existing conditions are manifestations of biological age, whereas aging and diseases are two sides of the same coin [24–26]. These conditions are typical age-related diseases: hypertension, diabetes, obesity, ischemic heart disease (IHD) and chronic obstructive pulmonary disease (COPD) and other diseases [9, 12–23].
Of course, not all (only some) patients with age-related diseases die from COVID-19. In other words, age-related diseases are necessary but not sufficient for mortality from COVID-19.
Age and pre-existing (age-related) diseases are interdependent. A number and severity of diseases correlate with age. An average 60 year old person has more age-related diseases than an average 50 your old person. Yet, a particular 60 year old person may have no age-related diseases, whereas a particular 50 year old person may have multiple diseases including hypertension, diabetes, obesity and cancer. In this case, it is a chronologically younger person who is biologically older. And it is the biological age that determines the likelihood of death from COVID-19.
Male Gender:
At the same age, the mortality rate is twice higher in men than in women [9, 27, 28], in part, because men age faster than women and, at any chronological age, men are biologically older than women [29].
So, three rules can be combined in one: COVID-19 vulnerability is determined by biological age. Biological age combines chronological age, age-related diseases and gender. A combination of all age-related diseases (and pre-diseases) is a biomarker of biological age. Figuratively, SARS-Cov-2 can “measure” biological age, which is thus the best predictor of mortality from both COVID-19 and other diseases.
Mortality from aging compared with COVID-19 mortality
Aging can be measured as an increase in the probability of death with age. Mortality increases exponentially, starting from age 8-9. Men have a higher “normal” age-related death rate than women because men age faster than women [29].
COVID-19 mortality rate parallels the “expected” aging-related death rate (Supplementary Figure 1) and see second graph in: https://medium.com/wintoncentre/how-much-normal-risk-does-covid-represent-4539118e1196.
Chances to die from COVID-19 are proportional to chances to die from aging itself at any age. The only discrepancy between natural and COVID-19 mortality is observed below the age of 8 years old. Whereas natural death rate is relatively high, COVID-19 mortality is low (no mortality [11]). This discrepancy will be discussed later. But first how do animals, including humans, die from aging?
Cytokine storm as a hyperfunction
Severe COVID-19 is characterized by hyper-inflammation, cytokine storm, acute respiratory distress syndrome (ARDS), damage to the lung, heart and kidneys [36–39].
In response to viral replication, hyperfunctional monocytes and macrophages infiltrate the lung, causing hyper-inflammation and hyper-secretion of cytokines such as interleukin (IL)-6, IL-2, IL-7, IL-1ra, interferon-γ inducible protein (IP)-10, tumor necrosis factor (TNF)-α, ferritin, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP) 1-α, granulocyte-colony stimulating factor (G-CSF), C-reactive protein (CRP) and procalcitonin. [22, 36–42].
This leads to leukocyte recruitment, vascular permeability, edema and further pulmonary damage in vicious cycle [37, 38, 41, 43, 44]. Hyper-inflammation becomes systemic, in turn causing hyper-coagulation and thrombosis, disseminated intravascular coagulation [45]. This causes injury of distant organs such as the kidneys. Pre-existing organ damage (late stages of age-related diseases) exacerbates organ damage caused by cytokine storm [42, 43, 46]. In addition, cellular hyper-functions and systemic hyper-inflammation may lead to cellular exhaustion, such as exhaustion of lymphocytes (lymphopenia) [47–49]. Hypercoagulation is associated with hyperactive fibrinolysis and increased D-dimer blood levels [23]. Cytokine storm is a systemic hyperfunctional response (Figure 1).
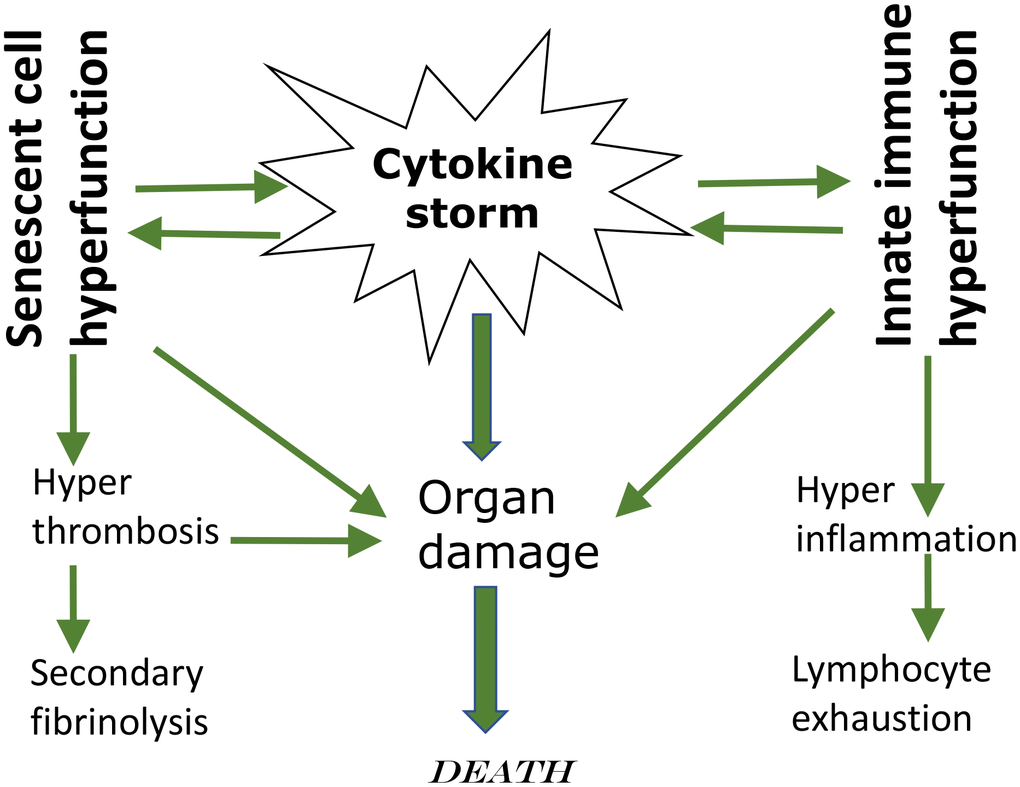
Figure 1. Cytokine storm as a systemic hyperfunction.
Of course, age-related hyperfunctional response, such as cytokine storm, is not caused by lifelong accumulation of molecular damage. Aging is not caused by molecular damage after all. Instead it’s a continuation of developmental/growth programs that lead to hyper-functions and in turn eventually to dysfunctions.
Hyperfunction theory of quasi-programmed aging
“Quasi” means “resembling” or “seemingly, but not really.” Quasi-program of aging is not a program but a continuation of developmental programs that were not switched off upon their completion [24, 50]. They purposelessly unfold, leading to age-related diseases, secondary organ failure and death. Quasi-programmed (program-like) aging is associated with higher than optimal cellular and systemic functions, which eventually, via cellular exhaustion and organ damage, lead to functional decline (Figure 2). For example, starting from birth, blood pressure increases and continues to increase after organismal growth is completed. Therefore, hypertension is the most prevalent age-related disease. In turn, hypertension can cause organ damage: stroke, infarction and renal failure. Similarly, obesity develops in post-development as a continuation of growth (yet, it can be prevented by low caloric diets, illustrating that quasi-program of aging can be decelerated).
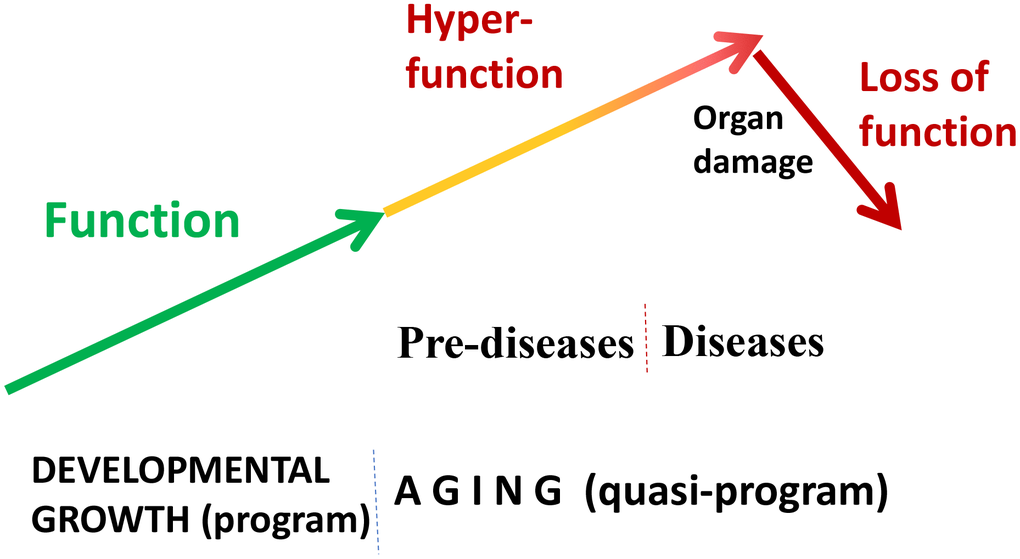
Figure 2. Quasi-programmed hyperfunctional aging. Aging is a continuation of developmental programs that were not switched off upon their completion. An increase in cellular and systemic functions (manifested as pre-diseases and then as diseases) leads to eventual organ damage and secondary loss of function.
Hyperfunction is an excessive normal cellular function: contraction by smooth muscle cells (SMC), adhesion and aggregation by blood platelets, insulin secretion by beta-cells, lipid accumulation by adipocytes, secretion by stromal and immune cells, oxidative burst by leukocytes, just to name a few. When higher than optimal, they cause vasoconstriction and hypertension, thrombosis, hyperinsulinemia, hypertrophy, hyperplasia, obesity, hyper-secretory phenotype or Senescence-associated secretory phenotype (SASP), hyper-inflammation and so on.
Hyper-function is not necessarily an absolutely increased function. It may be also insufficiently decreased function (relative hyperfunction). Levels of IGF-1 and growth hormone decrease during lifespan. Despite this decrease, IGF-1 levels are still higher than optimal (relative hyper-function) because further genetic decrease in IGF-1 levels (by genetic means) extends health span and lifespan in mammals [51–53].
Cellular hyperfunctions may eventually switch to cellular exhaustion and loss of functions at late stages. During the course of type II diabetes, mTOR overactivation and hyperinsulinemia eventually lead to beta-cell exhaustion and insulin insufficiency, from pre-diabetes to diabetes [54, 55]. As another example, after puberty, hyperstimulation of the ovary eventually leads to oocyte exhaustion and menopause (see Figure 3 in ref. [29]). Depletion of naïve lymphocytes is another example, as reviewed here later. Age-related alterations are mostly noticed when they switch to functional decline, which is a late event.

Figure 3. COVID-19 vulnerability as an age-related disease. Age-related diseases, including COVID-19 vulnerability, are manifestations of aging. Abbreviations: Ischemic heart disease (IHD); Chronic obstructive pulmonary disease (COPD).
In some cases, functional decline can be primary and programmed. For example, thymus involution (replacement of T cells by adipocytes) starts early in life, accelerates at puberty and continues later. Still loss of thymocytes and their niches may be in part due to adipocyte hyperplasia and hypertrophy [56]. In fact, obesity accelerates involution, whereas calorie restriction decelerates it [57, 58]. Furthermore, the oblation of sex hormones decelerates or even reverses thymus involution [59]. Thus, involution is triggered by adipocyte hyperplasia and increased production of sex hormones during puberty [56].
Quasi-programmed aging is not driven by molecular damage. It is driven by nutrient/hormone/cytokine-sensing and growth-promoting signaling pathways such as Target of Rapamycin (TOR; mTOR), which are involved in developmental growth and later cause hyperfunctional aging and its diseases [24, 26].
Inflamm-aging and immunosenescence
With hundreds of cell types acting in concert, the immune system is so complex that we cannot discuss age-related alterations without oversimplification. The most noticeable alteration is that memory T and B cells replace naive T and B cells [62]. (This seems natural since life-long exposure to pathogens replaces naïve cells by memory cells). Replacement of naïve immune cells decreases adaptive responses to novel antigens such as SARS-CoV-2. In contrast, immune protection by memory T cells from viral re-infection with known pathogens is usually increased with age [62].
Immune responses are roughly divided into (a) innate responses, carried mostly by neutrophils, macrophages and NK cells, which react to pathogen rapidly and nonspecifically, and (b) adaptive responses, carried by T and B lymphocytes, which are delayed, slower and specific (e.g., antigen-specific clonal expansion of T and B lymphocytes and antibody production by B lymphocytes) [63–65]. In the elderly, immune responses to SARS-CoV-1/2 are “stuck in innate immunity,” with insufficient progression to adaptive immunity [37]. However, decline in adaptive response, such as antibody production, plays little role in COVID-19 mortality. It is hyper-functional innate immunity, hyper-inflammation, cytokine storm and hyper-coagulation that lead to organ failure and death. In agreement, hyper inflammatory response rather than high virus numbers leads to death of SARS-CoV-infected old nonhuman primates [66].
Aging is associated with diseases of immune hyper-function such as autoimmune disorders with paradoxical increase in certain signaling pathways and cytokine levels [67–69].
In the elderly, innate immune cells are in a state of sustained activation, producing pro-inflammatory cytokines [67, 70–72]. Increased pro-inflammatory activity by the innate immune system, especially by monocytes/macrophages, is a state of alertness and hyper-reactivity on the cost of potential age-related inflammatory diseases [67, 70–72]. Whereas some functions are decreased, others are increased. According to the inflamm-aging concept, innate immune system overtakes adaptive immune system in aging. Cause-effect relationships are bi-directional: immunosenescence (namely, a decrease in adaptive response) is a cause and consequence of inflamm-aging [67, 70–72].
We can consider inflamm-aging as an example of hyper-function. While some functions are decreased, others are increased. Hyper-function is damaging. (In analogy, increased electric power, without an adaptor, would damage a laptop). Damaging hyper-functions can lead to loss of function and cellular exhaustion. And vice versa, loss of function may cause compensatory hyper-functions of another components.
Cellular senescence as a continuation of growth
Cellular senescence is a continuation of cellular growth, when actual growth is completed [73, 74]. In proliferating cells, cellular mass growth is balanced by cell division. Cells grow in size and then divide. When the cell cycle is blocked (e.g., p21 and p16), then growth-promoting pathways such as mTOR and MAPK drive conversion to senescence (geroconversion) [24, 74, 75]. During geroconversion, cells become hypertrophic and “fat”. Cellular functions increase: hyper-secretion and lysosomal hyper-function are manifested by SASP and beta-Gal staining. Hyper-activated growth-promoting pathways cause compensatory resistance to growth factors/insulin, permanent loss of re-proliferative potential [74]. Rapamycin, everolimus, pan-mTOR and MAPK inhibitors slows down geroconversion, maintaining reversible quiescence instead of senescence [73, 76–88].
Geroconversion is a continuation of cellular growth [73, 74]. Similarly, aging is a continuation of developmental growth (see Figure 1 in ref. [89]). When the developmental program is completed, it becomes a quasi-program of aging. As discussed in detail, chronically activated nutrient-sensing and growth-promoting pathways drive age-related diseases, culminating in organismal death [24, 26].
Age-related diseases are quasi-programmed. Aging is a common cause of age-related diseases, a sum of all age-related diseases. They are diseases of hyper-function, secondary hypo-function and compensation reactions [25]; they are deadly manifestations of aging.
From activation of cellular functions to systemic hyperfunctions, from diseases to organ damage and death, hyperfunction theory of quasi-programmed aging describes the sequence of events [26]. And as discussed in 2006, suppression of aging by gero-suppressants, such as rapamycin, will prevent and treat all age-related diseases [24]. This point of view is becoming widely accepted and, in recent literature, quasi-programmed model of diseases (2006) is called “geroscience hypothesis” [2, 90].
Figuratively, rapamycin rejuvenates immunity [91].
If aging were functional decline due to accumulation of molecular damage, then it would be near to impossible to restore functions and rejuvenate the immune system. In contrast, if functional decline is secondary to hyperfunctions (see Figure 2 in ref. [89]), these hyperfunctions can be suppressed pharmacologically to restore lost functions. Typical drugs are inhibitors of their targets, rather than activators, so they decrease functions of their targets. By decreasing hyper-functions, which otherwise lead to secondary loss of functions, rapamycin may restore “lost” functions (Figure 4).
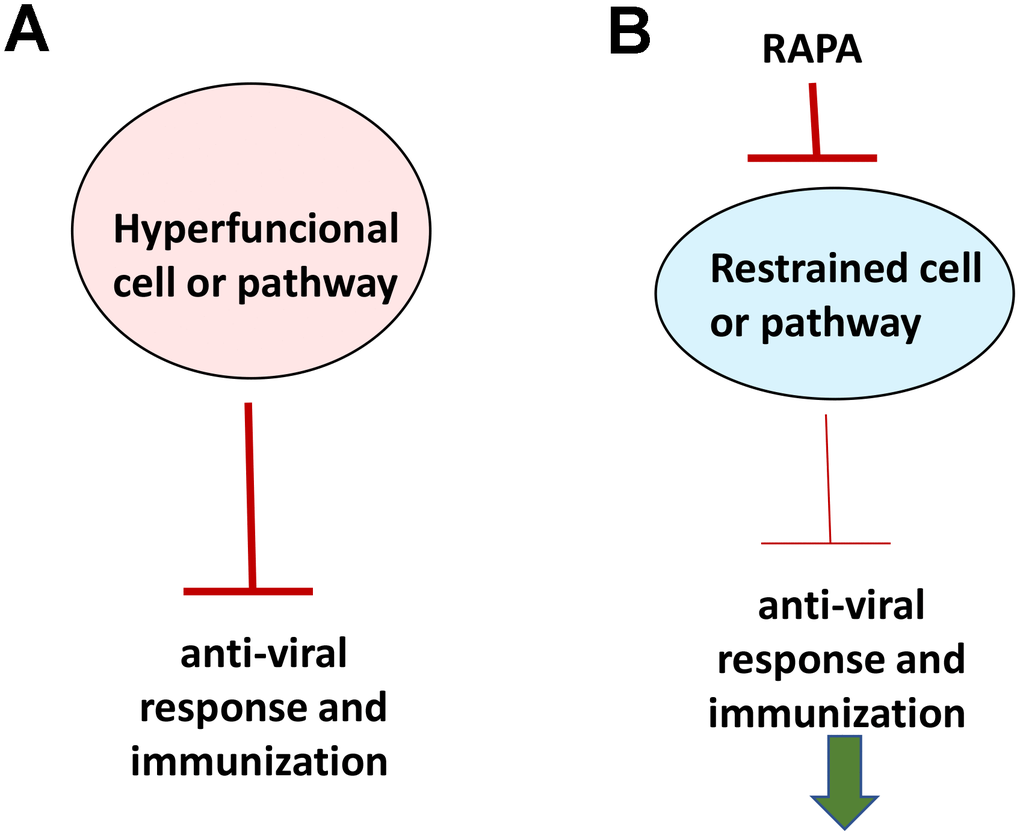
Figure 4. Rejuvenating immunity by inhibiting hyperfunction. (A) Specific hyper-functional cells (or signaling pathways) can inhibit some other cell types (or pathways) that are needed for proper anti-viral response and immunization. (B) By inhibiting hyper-functional cells or pathways, rapamycin can reactivate “loss-of-function” otherwise suppressed by hyper-functional cells or pathways.
Rapamycin improves vaccination against viruses such as influenza in old mice, monkeys and humans [92–100]. Importantly, rapamycin increases pathogen-specific but not graft-reactive CD8+ T cell responses [95, 101]. Therefore, rapamycin and everolimus can both be used to prevent donor organ rejection and improve adaptive immunity against new pathogens [96].
Differentiation is an increase of tissue-specific cellular functions. Terminally differentiated B, T, and NK cells can rapidly react to already known pathogens [102]. Decrease in naïve T and B lymphocytes (and thus diminished response to novel antigens) results in part from cellular hyper-differentiation in the immune system [64, 103]. Hyper-functional differentiation can be counteracted by rapamycin [98].
As another example, age-related exhaustion of stem cells is partially due to loss of quiescence caused by growth over-stimulation [92, 104–106]. In general, senescent cells characterized by hyper-proliferative drive coupled with cell cycle arrest [77]. In young mice, mTOR hyper-activation causes senescence of hematopoietic stem cells (HSC) and decreases lymphopoiesis [92]. In old mice, rapamycin rejuvenates hematopoiesis, and improves vaccination against influenza virus [92].
Third, production of lymphoid cells may be decreased because of disruption of hypoxic niches due to adipocytes hyperplasia [56, 107]. Hypoxic niches can preserve HSC [108, 109] probably because hypoxia inhibits mTOR and cellular senescence [110]. In agreement, rapamycin preserves HSCs [92, 98, 111, 112] reduces the proportion of memory cells and maintains a pool of naïve T cells [92, 98].
Fourth, growth factor (GF)- and insulin-resistance is loss of function because cells cannot respond to GF/insulin. But it may be caused by over-activated mTOR, which via S6K/IRS feedback loop blocks insulin and GF signaling. Rapamycin abrogates the loop restoring signaling [113–118].
Anti-aging medicine
A high prevalence of age-related diseases, often called “diseases of civilization,” is a success story of modern medicine. In the past, most people did not live long enough to develop age-related diseases and those who developed them died soon after. Due to medical advances, people survive to 85 on average, despite suffering from age-related diseases. Standard medicine preferentially extends life span, without necessarily affecting health span (see Figure 3 in ref. [119]). For example, defibrillation and coronary stenting can save life but not cure heart disease. It is anti-aging interventions that extend health span, delaying diseases, thus extending lifespan. Aging is a common cause of all age-related diseases. By suppressing aging, anti-aging interventions may delay all age-related diseases [119].
As a well-known example, low calorie diets such as calorie restriction, intermittent fasting, and low carbohydrate diets extend both health and lifespan. Figuratively, low calorie diets prolong life by improving health. Nutrients and obesity activate growth-promoting pathways (e.g., mTOR), thus accelerating development of quasi-programmed (age-related) diseases. Obesity is associated with all age-related diseases from cancer to Alzheimer’s and from diabetes to sarcopenia. COVID-19 vulnerability is also associated with obesity [9, 19, 20, 22]. According to hyperfunction theory, obesity accelerates aging and all age-related conditions including COVID-19 vulnerability.
Diabetes is one of main risk factors of death in COVID-19 [5, 6, 12, 13, 15, 21]. Can type 2 diabetes, an age-related disease, be reversed? In remarkable studies, it was shown that a brief course (6-8 weeks) of very low calorie diets (VLCDs) can reverse type II diabetes. In one study, VLCD reversed diabetes in 46% of patients with up to a 6-year history of diabetes [120]. VLCD is most effective for its prevention and at early stages of diabetes [121]. This anti-aging modality is so simple that remission can be achieved at home by health-motivated individuals [122]. Simultaneously, it treats other age-related diseases such hypertension [123]. Obesity is associated with other diseases of hyperfunction from diabetes and sarcopenia to cancer and Alzheimer’s’ disease. Since age-related diseases are predictors of COVID-19 mortality, VLCD in theory may decrease COVID-19 vulnerability.
Rapamycin and everolimus as anti-aging drugs
In the soil of Easter Island, a complex bacteria produces anti-fungal antibiotic rapamycin to suppress yeast growth but, as a by-product, it also suppresses yeast aging (quasi-programed aging is a continuation of growth). Approved for human use in 1999, Rapamycin (Sirolimus) and its close analog Everolimus are widely used in several diseases including cancer and organ transplantation. Hundreds of clinical trials (and twenty years of clinical practice) have ensured their safety and good tolerability especially in healthy older adults [119].
Currently, several anti-aging clinics prescribe rapamycin out of label to prevent age-related diseases and slow aging. Hundreds of recent reviews discussed rapamycin and everolimus in detail, so I will just emphasize a few points:
Crucial prediction of hyper-function theory of quasi-programmed aging in 2006 was that rapamycin will slow aging, extend healthspan and lifespan and decrease all age-related [124]. It has been confirmed: it extends lifespan in animals from worm to mammals. In some strains of short-lived mutant mice, it extends life span two fold [98, 125].
Rapamycin slows geroconversion to cellular senescence in cell culture [74].
mTOR is a potential therapeutic target in chronic obstructive pulmonary disease COPD [126], [127]. Rapamycin (sirolimus) is already approved and successfully used in lymphangioleiomyomatosis (LAM), a progressive, cystic lung disease, associated with inappropriate activation of mTOR [128]. Long-term daily use of rapamycin improves lung function without causing serious side effects (and of course no even minor side effects in the lung, given that rapamycin improves lung function) [128].
Despite widespread misunderstanding, rapamycin and everolimus do not cause diabetes. In contrast, they prevent diabetic complications in animals with diabetes (see for references [129]). In rodents, in some conditions they may cause symptoms of starvation pseudo-diabetes similar to prolong fasting and ketogenic diet [129]. Although, the Johnson study found a slight but significant correlation between Medicare billing for insulin and the use of rapamycin in renal transplant patients, this correlation was mechanistically explained by interaction of rapamycin with two other drugs used in the same patients [130, 131]. In cancer patients, everolimus may cause reversible hyperglycemia as a mild, infrequent and reversible side effect after several weeks of daily high doses of everolimus and rapamycin [132]. Mechanistically, everolimus decrease insulin production, not causing insulin resistance [132]. If anything, everolimus and rapamycin can be considered to treat complications of type II diabetes and prevent hyperinsulinemia and obesity ([129] and references within). What actually contributes to type 2 diabetes is excess of nutrients (and especially carbohydrates), which activate mTOR and cause hyperinsulinemia and insulin resistance.
Potential applications of rapamycin/everolimus to COVID-19
As soon as COVID-19 epidemic started, it become clear that COVID-19 vulnerability is an aging-dependent condition and the use of rapamycin (Sirolimus) was immediately suggested by independent researchers [1, 3, 133–137]. These proposals were based on a mixture of several rationales, which need to be clearly distinguished. In theory, there are at least three independent applications of rapamycin and everolimus for COVID-19. Currently, they all are still hypothetical.
Anti-aging effect (Figure 5). By decreasing biological age and preventing age-related diseases, a long-term rapamycin therapy may in theory decrease COVID-19 mortality rate in the elderly. Anti-aging application is especially important because it is beneficial regardless of COVID-19. After all, mortality rate from aging and its diseases is 100%, causing more than 2 million deaths in the USA annually. Continuous use of rapamycin is expected to improve health, decrease age-related diseases and extend healthy lifespan, rendering individuals less vulnerable, when infected with the virus.
Rejuvenating immunity. As we discussed in section “Figuratively, rapamycin rejuvenates immunity” [91], mTOR inhibitors can improve immunity to viral infections, improve immunization and vaccination to some viruses such as flu [92–100, 111, 112, 138]. In addition, viruses such as flu [139] and coronavirus (MERS-CoV) [140] depend on mTOR activity for replication. Currently, however, there are no data regarding COVID-19. Although aimed to evaluate safety, Phase 1 clinical trial “Sirolimus in COVID-19 Phase 1 (SirCO-1)” may reveal anti-viral effects too https://clinicaltrials.gov/ct2/show/NCT04371640.
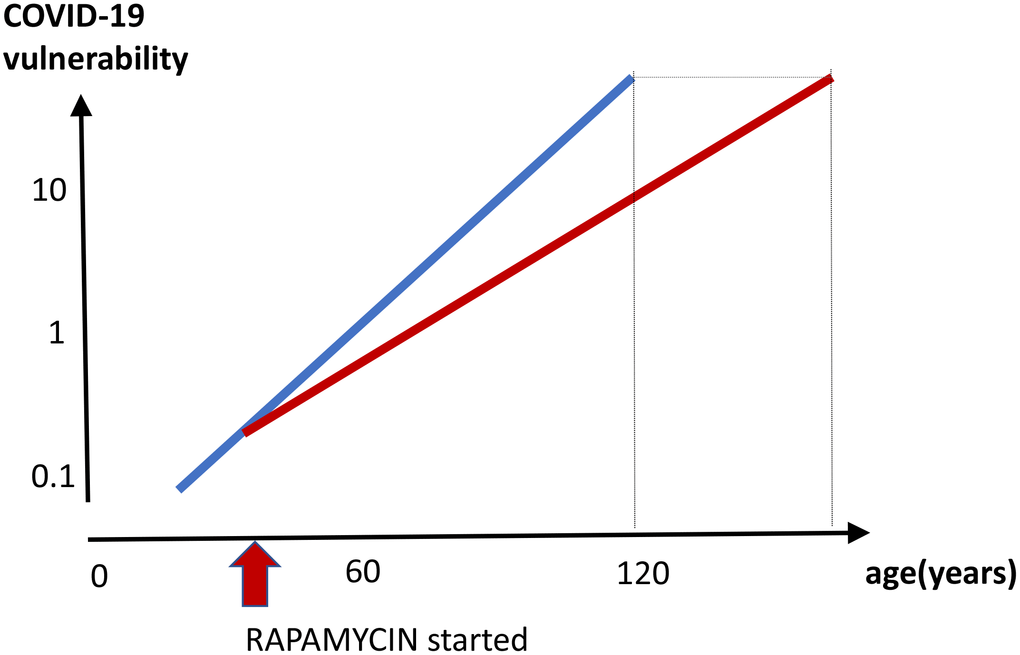
Figure 5. Prevention of COVID-19 vulnerability by staying young. Hypothetical graph in the absence of COVID-19. COVID-19 vulnerability (log scale) increases exponentially with age (blue line). The line ends at age 120, a maximum recorded age for humans. In theory, a continuous rapamycin treatment would slow down an increase of the vulnerability with age (red line). The increase is still logarithmic but at a different slope, because rapamycin slows the aging process. The maximum lifespan, in the absence of COVID-19, is extended because the 100% natural death threshold is achieved later.
3. Potential suppression of cytokine storm and hyper-inflammation (Figure 1). As we discussed in the section “Cytokine storm is a hyperfunction”, cytokine storm and hyper-inflammation is a main cause of death in COVID-19 pneumonia [36–40, 42, 45, 135, 141–143] Rapamycin, an anti-inflammatory agent, inhibits hyper-functions, cellular senescence and decrease secretion of cytokines ([74, 81, 144]. Rapamycin inhibits the Jak2/Stat4 signaling pathway [145] and reduces IF-γ and TNF-α levels [112]. Rapamycin (Sirolimus) treatment improves outcomes in patients with severe H1N1 pneumonia and acute respiratory failure and was associated with improvement in virus clearance, and shortened ventilator days [146]. Clinical trial “Sirolimus Treatment in Hospitalized Patients With COVID-19 Pneumonia (SCOPE)” has been started https://clinicaltrials.gov/ct2/show/NCT04341675.
Disclaimer
This review is intended for a professional audience, to stimulate new ideas and to aid the global efforts to develop effective treatments for COVID-19 disease. This article does not represent medical advice or recommendations to patients. The media should exercise caution and seek expert medical advice for interpretation, when referring to this article.
Supplementary Materials
Conflicts of Interest
The author declares no conflicts of interest.
References
- 1. Zhavoronkov A. Geroprotective and senoremediative strategies to reduce the comorbidity, infection rates, severity, and lethality in gerophilic and gerolavic infections. Aging (Albany NY). 2020; 12:6492–510. https://doi.org/10.18632/aging.102988 [PubMed]
- 2. Promislow DE. A geroscience perspective on COVID-19 mortality. J Gerontol A Biol Sci Med Sci. 2020. [Epub ahead of print]. https://doi.org/10.1093/gerona/glaa094 [PubMed]
- 3. Sargiacomo C, Sotgia F, Lisanti MP. COVID-19 and chronological aging: senolytics and other anti-aging drugs for the treatment or prevention of corona virus infection? Aging (Albany NY). 2020; 12:6511–17. https://doi.org/10.18632/aging.103001 [PubMed]
- 4. Leung C. Risk factors for predicting mortality in elderly patients with COVID-19: a review of clinical data in China. Mech Ageing Dev. 2020; 188:111255. https://doi.org/10.1016/j.mad.2020.111255 [PubMed]
- 5. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in wuhan, China: a retrospective cohort study. Lancet. 2020; 395:1054–62. https://doi.org/10.1016/S0140-6736(20)30566-3 [PubMed]
- 6. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, Huang H, Zhang L, Zhou X, Du C, Zhang Y, Song J, Wang S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in wuhan, China. JAMA Intern Med. 2020. [Epub ahead of print]. https://doi.org/10.1001/jamainternmed.2020.0994 [PubMed]
- 7. Lauc G, Sinclair D. Biomarkers of biological age as predictors of COVID-19 disease severity. Aging (Albany NY). 2020; 12:6490–91. https://doi.org/10.18632/aging.103052 [PubMed]
- 8. Du RH, Liang LR, Yang CQ, Wang W, Cao TZ, Li M, Guo GY, Du J, Zheng CL, Zhu Q, Hu M, Li XY, Peng P, Shi HZ. Predictors of mortality for patients with COVID-19 pneumonia caused by SARS-CoV-2: a prospective cohort study. Eur Respir J. 2020; 55:2000524. https://doi.org/10.1183/13993003.00524-2020 [PubMed]
- 9. Palaiodimos L, Kokkinidis DG, Li W, Karamanis D, Ognibene J, Arora S, Southern WN, Mantzoros CS. Severe obesity, increasing age and male sex are independently associated with worse in-hospital outcomes, and higher in-hospital mortality, in a cohort of patients with COVID-19 in the bronx, new york. Metabolism. 2020; 108:154262. https://doi.org/10.1016/j.metabol.2020.154262 [PubMed]
- 10. Banerjee A, Pasea L, Harris S, Gonzalez-Izquierdo A, Torralbo A, Shallcross L, Noursadeghi M, Pillay D, Sebire N, Holmes C, Pagel C, Wong WK, Langenberg C, et al. Estimating excess 1-year mortality associated with the COVID-19 pandemic according to underlying conditions and age: a population-based cohort study. Lancet. 2020; 395:1715–25. https://doi.org/10.1016/S0140-6736(20)30854-0 [PubMed]
- 11. Lu Q, Shi Y. Coronavirus disease (COVID-19) and neonate: what neonatologist need to know. J Med Virol. 2020; 10:1002. https://doi.org/10.1002/jmv.25740 [PubMed]
- 12. Hussain A, Bhowmik B, do Vale Moreira NC. COVID-19 and diabetes: knowledge in progress. Diabetes Res Clin Pract. 2020; 162:108142. https://doi.org/10.1016/j.diabres.2020.108142 [PubMed]
- 13. Huang I, Lim MA, Pranata R. Diabetes mellitus is associated with increased mortality and severity of disease in COVID-19 pneumonia - a systematic review, meta-analysis, and meta-regression. Diabetes Metab Syndr. 2020; 14:395–403. https://doi.org/10.1016/j.dsx.2020.04.018 [PubMed]
- 14. Mehra MR, Desai SS, Kuy S, Henry TD, Patel AN. Cardiovascular disease, drug therapy, and mortality in covid-19. N Engl J Med. 2020. [Epub ahead of print]. https://doi.org/10.1056/NEJMoa2007621 [PubMed]
- 15. Yan Y, Yang Y, Wang F, Ren H, Zhang S, Shi X, Yu X, Dong K. Clinical characteristics and outcomes of patients with severe covid-19 with diabetes. BMJ Open Diabetes Res Care. 2020; 8:e001343. https://doi.org/10.1136/bmjdrc-2020-001343 [PubMed]
- 16. Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, Madhur MS, Tomaszewski M, Maffia P, D’Acquisto F, Nicklin SA, Marian AJ, Nosalski R, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020. [Epub ahead of print]. https://doi.org/10.1093/cvr/cvaa106 [PubMed]
- 17. Hu J, Zhang X, Zhang X, Zhao H, Lian J, Hao S, Jia H, Yang M, Lu Y, Xiang D, Cai H, Zhang S, Gu J, et al. COVID-19 patients with hypertension have more severity condition, and ACEI/ARB treatment have no infulence on the clinical severity and outcome. J Infect. 2020; S0163-4453:30334–30. https://doi.org/10.1016/j.jinf.2020.05.056 [PubMed]
- 18. Zhang J, Wu J, Sun X, Xue H, Shao J, Cai W, Jing Y, Yue M, Dong C. Association of hypertension with the severity and fatality of SARS-CoV-2 infection: a meta-analysis. Epidemiol Infect. 2020; 148:e106. https://doi.org/10.1017/S095026882000117X [PubMed]
- 19. Dietz W, Santos-Burgoa C. Obesity and its implications for COVID-19 mortality. Obesity (Silver Spring). 2020; 28:1005. https://doi.org/10.1002/oby.22818 [PubMed]
- 20. Zhang F, Xiong Y, Wei Y, Hu Y, Wang F, Li G, Liu K, Du R, Wang CY, Zhu W. Obesity predisposes to the risk of higher mortality in young COVID-19 patients. J Med Virol. 2020. [Epub ahead of print]. https://doi.org/10.1002/jmv.26039 [PubMed]
- 21. Wang B, Li R, Lu Z, Huang Y. Does comorbidity increase the risk of patients with COVID-19: evidence from meta-analysis. Aging (Albany NY). 2020; 12:6049–57. https://doi.org/10.18632/aging.103000 [PubMed]
- 22. Korakas E, Ikonomidis I, Kousathana F, Balampanis K, Kountouri A, Raptis A, Palaiodimou L, Kokkinos A, Lambadiari V. Obesity and COVID-19: immune and metabolic derangement as a possible link to adverse clinical outcomes. Am J Physiol Endocrinol Metab. 2020. [Epub ahead of print]. https://doi.org/10.1152/ajpendo.00198.2020 [PubMed]
- 23. Ji HL, Zhao R, Matalon S, Matthay MA. Elevated plasmin(ogen) as a common risk factor for COVID-19 susceptibility. Physiol Rev. 2020; 100:1065–75. https://doi.org/10.1152/physrev.00013.2020 [PubMed]
- 24. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5:2087–102. https://doi.org/10.4161/cc.5.18.3288 [PubMed]
- 25. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging (Albany NY). 2009; 1:281–88. https://doi.org/10.18632/aging.100034 [PubMed]
- 26. Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012; 181:1142–46. https://doi.org/10.1016/j.ajpath.2012.06.024 [PubMed]
- 27. Gebhard C, Regitz-Zagrosek V, Neuhauser HK, Morgan R, Klein SL. Impact of sex and gender on COVID-19 outcomes in europe. Biol Sex Differ. 2020; 11:29. https://doi.org/10.1186/s13293-020-00304-9 [PubMed]
- 28. Jin JM, Bai P, He W, Wu F, Liu XF, Han DM, Liu S, Yang JK. Gender differences in patients with COVID-19: focus on severity and mortality. Front Public Health. 2020; 8:152. https://doi.org/10.3389/fpubh.2020.00152 [PubMed]
- 29. Blagosklonny MV. Why men age faster but reproduce longer than women: mTOR and evolutionary perspectives. Aging (Albany NY). 2010; 2:265–73. https://doi.org/10.18632/aging.100149 [PubMed]
- 30. Wang H, Zhang Z, Gems D. Monsters in the uterus: teratoma-like tumors in senescent C. Elegans result from a parthenogenetic quasi-program. Aging (Albany NY). 2018; 10:1188–89. https://doi.org/10.18632/aging.101486 [PubMed]
- 31. Xi J, Cai J, Cheng Y, Fu Y, Wei W, Zhang Z, Zhuang Z, Hao Y, Lilly MA, Wei Y. The TORC1 inhibitor Nprl2 protects age-related digestive function in Drosophila. Aging (Albany NY). 2019; 11:9811–28. https://doi.org/10.18632/aging.102428 [PubMed]
- 32. Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?”. Aging (Albany NY). 2012; 4:861–77. https://doi.org/10.18632/aging.100525 [PubMed]
- 33. Corrada MM, Brookmeyer R, Paganini-Hill A, Berlau D, Kawas CH. Dementia incidence continues to increase with age in the oldest old: the 90+ study. Ann Neurol. 2010; 67:114–21. https://doi.org/10.1002/ana.21915 [PubMed]
- 34. Blagosklonny MV. Disease or not, aging is easily treatable. Aging (Albany NY). 2018; 10:3067–78. https://doi.org/10.18632/aging.101647 [PubMed]
- 35. Williams EJ, Embleton ND, Bythell M, Ward Platt MP, Berrington JE. The changing profile of infant mortality from bacterial, viral and fungal infection over two decades. Acta Paediatr. 2013; 102:999–1004. https://doi.org/10.1111/apa.12341 [PubMed]
- 36. Akhmerov A, Marbán E. COVID-19 and the heart. Circ Res. 2020; 126:1443–55. https://doi.org/10.1161/CIRCRESAHA.120.317055 [PubMed]
- 37. Nikolich-Zugich J, Knox KS, Rios CT, Natt B, Bhattacharya D, Fain MJ. SARS-CoV-2 and COVID-19 in older adults: what we may expect regarding pathogenesis, immune responses, and outcomes. Geroscience. 2020; 42:505–14. https://doi.org/10.1007/s11357-020-00186-0 [PubMed]
- 38. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘Cytokine storm’ in COVID-19. J Infect. 2020; 80:607–13. https://doi.org/10.1016/j.jinf.2020.03.037 [PubMed]
- 39. Henderson LA, Canna SW, Schulert GS, Volpi S, Lee PY, Kernan KF, Caricchio R, Mahmud S, Hazen MM, Halyabar O, Hoyt KJ, Han J, Grom AA, et al. On the alert for cytokine storm: immunopathology in COVID-19. Arthritis Rheumatol. 2020; 10:1002. https://doi.org/10.1002/art.41285 [PubMed]
- 40. Yao Z, Zheng Z, Wu K, Junhua Z. Immune environment modulation in pneumonia patients caused by coronavirus: SARS-CoV, MERS-CoV and SARS-CoV-2. Aging (Albany NY). 2020; 12:7639–51. https://doi.org/10.18632/aging.103101 [PubMed]
- 41. Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F, Wang F, Li G, Li Y, Xing L, Peng L, Yang M, Cao M, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol. 2020; S0091-6749:30576–75. https://doi.org/10.1016/j.jaci.2020.04.027 [PubMed]
- 42. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. 2020; 19:102537. https://doi.org/10.1016/j.autrev.2020.102537 [PubMed]
- 43. Vaninov N. In the eye of the COVID-19 cytokine storm. Nat Rev Immunol. 2020; 20:277. https://doi.org/10.1038/s41577-020-0305-6 [PubMed]
- 44. Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun. 2020; 109:102433. https://doi.org/10.1016/j.jaut.2020.102433 [PubMed]
- 45. Dolhnikoff M, Duarte-Neto AN, de Almeida Monteiro RA, da Silva LF, de Oliveira EP, Saldiva PH, Mauad T, Negri EM. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost. 2020; 18:1517–19. https://doi.org/10.1111/jth.14844 [PubMed]
- 46. Siddiqi HK, Mehra MR. COVID-19 illness in native and immunosuppressed states: a clinical-therapeutic staging proposal. J Heart Lung Transplant. 2020; 39:405–07. https://doi.org/10.1016/j.healun.2020.03.012 [PubMed]
- 47. Fathi N, Rezaei N. Lymphopenia in COVID-19: therapeutic opportunities. Cell Biol Int. 2020. [Epub ahead of print]. https://doi.org/10.1002/cbin.11403 [PubMed]
- 48. Cao X. COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol. 2020; 20:269–70. https://doi.org/10.1038/s41577-020-0308-3 [PubMed]
- 49. Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang YQ, Wang Q, Miao H. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct Target Ther. 2020; 5:33. https://doi.org/10.1038/s41392-020-0148-4 [PubMed]
- 50. Blagosklonny MV. Aging is not programmed: genetic pseudo-program is a shadow of developmental growth. Cell Cycle. 2013; 12:3736–42.
- 51. Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ. The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol. 2013; 9:366–76. https://doi.org/10.1038/nrendo.2013.67 [PubMed]
- 52. Bartke A, List EO, Kopchick JJ. The somatotropic axis and aging: benefits of endocrine defects. Growth Horm IGF Res. 2016; 27:41–45. https://doi.org/10.1016/j.ghir.2016.02.002 [PubMed]
- 53. Junnila RK, Duran-Ortiz S, Suer O, Sustarsic EG, Berryman DE, List EO, Kopchick JJ. Disruption of the GH receptor gene in adult mice increases maximal lifespan in females. Endocrinology. 2016; 157:4502–13. https://doi.org/10.1210/en.2016-1649 [PubMed]
- 54. Guillén C, Benito M. mTORC1 overactivation as a key aging factor in the progression to type 2 diabetes mellitus. Front Endocrinol (Lausanne). 2018; 9:621. https://doi.org/10.3389/fendo.2018.00621 [PubMed]
- 55. Yuan T, Rafizadeh S, Gorrepati KD, Lupse B, Oberholzer J, Maedler K, Ardestani A. Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia. 2017; 60:668–78. https://doi.org/10.1007/s00125-016-4188-9 [PubMed]
- 56. Chinn IK, Blackburn CC, Manley NR, Sempowski GD. Changes in primary lymphoid organs with aging. Semin Immunol. 2012; 24:309–20. https://doi.org/10.1016/j.smim.2012.04.005 [PubMed]
- 57. Yang H, Youm YH, Vandanmagsar B, Rood J, Kumar KG, Butler AA, Dixit VD. Obesity accelerates thymic aging. Blood. 2009; 114:3803–12. https://doi.org/10.1182/blood-2009-03-213595 [PubMed]
- 58. Yang H, Youm YH, Dixit VD. Inhibition of thymic adipogenesis by caloric restriction is coupled with reduction in age-related thymic involution. J Immunol. 2009; 183:3040–52. https://doi.org/10.4049/jimmunol.0900562 [PubMed]
- 59. Hince M, Sakkal S, Vlahos K, Dudakov J, Boyd R, Chidgey A. The role of sex steroids and gonadectomy in the control of thymic involution. Cell Immunol. 2008; 252:122–38. https://doi.org/10.1016/j.cellimm.2007.10.007 [PubMed]
- 60. Ramsamy K, Subramaniyan R, Patra AK. An observational study of the association between androgenetic alopecia and size of the prostate. Int J Trichology. 2016; 8:62–66. https://doi.org/10.4103/0974-7753.188034 [PubMed]
- 61. Mannino DM, Thorn D, Swensen A, Holguin F. Prevalence and outcomes of diabetes, hypertension and cardiovascular disease in COPD. Eur Respir J. 2008; 32:962–69. https://doi.org/10.1183/09031936.00012408 [PubMed]
- 62. Davenport B, Eberlein J, Nguyen TT, Victorino F, Jhun K, Abuirqeba H, van der Heide V, Heeger P, Homann D. Aging boosts antiviral CD8+T cell memory through improved engagement of diversified recall response determinants. PLoS Pathog. 2019; 15:e1008144. https://doi.org/10.1371/journal.ppat.1008144 [PubMed]
- 63. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O’Neill LA, Xavier RJ. Trained immunity: a program of innate immune memory in health and disease. Science. 2016; 352:aaf1098. https://doi.org/10.1126/science.aaf1098 [PubMed]
- 64. Pereira BI, Akbar AN. Convergence of innate and adaptive immunity during human aging. Front Immunol. 2016; 7:445. https://doi.org/10.3389/fimmu.2016.00445 [PubMed]
- 65. Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. 2018; 19:10–19. https://doi.org/10.1038/s41590-017-0006-x [PubMed]
- 66. Smits SL, de Lang A, van den Brand JM, Leijten LM, van IJcken WF, Eijkemans MJ, van Amerongen G, Kuiken T, Andeweg AC, Osterhaus AD, Haagmans BL. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010; 6:e1000756. https://doi.org/10.1371/journal.ppat.1000756 [PubMed]
- 67. Fulop T, Dupuis G, Baehl S, Le Page A, Bourgade K, Frost E, Witkowski JM, Pawelec G, Larbi A, Cunnane S. From inflamm-aging to immune-paralysis: a slippery slope during aging for immune-adaptation. Biogerontology. 2016; 17:147–57. https://doi.org/10.1007/s10522-015-9615-7 [PubMed]
- 68. Montgomery RR, Shaw AC. Paradoxical changes in innate immunity in aging: recent progress and new directions. J Leukoc Biol. 2015; 98:937–43. https://doi.org/10.1189/jlb.5MR0315-104R [PubMed]
- 69. Rostamzadeh D, Yousefi M, Haghshenas MR, Ahmadi M, Dolati S, Babaloo Z. mTOR signaling pathway as a master regulator of memory CD8+ t-cells, Th17, and NK cells development and their functional properties. J Cell Physiol. 2019; 234:12353–68. https://doi.org/10.1002/jcp.28042 [PubMed]
- 70. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000; 908:244–54. https://doi.org/10.1111/j.1749-6632.2000.tb06651.x [PubMed]
- 71. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, Witkowski JM, Franceschi C. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. 2018; 8:1960. https://doi.org/10.3389/fimmu.2017.01960 [PubMed]
- 72. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014 (Suppl 1); 69:S4–9. https://doi.org/10.1093/gerona/glu057 [PubMed]
- 73. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7:3355–61. https://doi.org/10.4161/cc.7.21.6919 [PubMed]
- 74. Blagosklonny MV. Rapamycin, proliferation and geroconversion to senescence. Cell Cycle. 2018; 17:2655–65. https://doi.org/10.1080/15384101.2018.1554781 [PubMed]
- 75. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4:358–62. https://doi.org/10.1038/sj.embor.embor806 [PubMed]
- 76. Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009; 8:1896–900. https://doi.org/10.4161/cc.8.12.8809 [PubMed]
- 77. Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013; 20:1241–49. https://doi.org/10.1038/cdd.2013.86 [PubMed]
- 78. Sousa-Victor P, García-Prat L, Muñoz-Cánoves P. Dual mTORC1/C2 inhibitors: gerosuppressors with potential anti-aging effect. Oncotarget. 2015; 6:23052–54. https://doi.org/10.18632/oncotarget.5563 [PubMed]
- 79. Leontieva OV, Blagosklonny MV. Gerosuppression by pan-mTOR inhibitors. Aging (Albany NY). 2016; 8:3535–51. https://doi.org/10.18632/aging.101155 [PubMed]
- 80. Walters HE, Deneka-Hannemann S, Cox LS. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY). 2016; 8:231–44. https://doi.org/10.18632/aging.100872 [PubMed]
- 81. Wang R, Yu Z, Sunchu B, Shoaf J, Dang I, Zhao S, Caples K, Bradley L, Beaver LM, Ho E, Löhr CV, Perez VI. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017; 16:564–74. https://doi.org/10.1111/acel.12587 [PubMed]
- 82. Gu Z, Tan W, Ji J, Feng G, Meng Y, Da Z, Guo G, Xia Y, Zhu X, Shi G, Cheng C. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY). 2016; 8:1102–14. https://doi.org/10.18632/aging.100925 [PubMed]
- 83. Kochetkova EY, Blinova GI, Bystrova OA, Martynova MG, Pospelov VA, Pospelova TV. Targeted elimination of senescent ras-transformed cells by suppression of MEK/ERK pathway. Aging (Albany NY). 2017; 9:2352–75. https://doi.org/10.18632/aging.101325 [PubMed]
- 84. Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012; 11:2391–401. https://doi.org/10.4161/cc.20683 [PubMed]
- 85. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA. 2010; 107:9660–64. https://doi.org/10.1073/pnas.1002298107 [PubMed]
- 86. Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci USA. 2014; 111:8832–37. https://doi.org/10.1073/pnas.1405723111 [PubMed]
- 87. Horvath S, Lu AT, Cohen H, Raj K. Rapamycin retards epigenetic ageing of keratinocytes independently of its effects on replicative senescence, proliferation and differentiation. Aging (Albany NY). 2019; 11:3238–49. https://doi.org/10.18632/aging.101976 [PubMed]
- 88. Cho S, Hwang ES. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol Cells. 2012; 33:597–604. https://doi.org/10.1007/s10059-012-0042-1 [PubMed]
- 89. Blagosklonny MV. Does rapamycin slow down time? Oncotarget. 2018; 9:30210–12. https://doi.org/10.18632/oncotarget.25788 [PubMed]
- 90. Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, et al. Geroscience: linking aging to chronic disease. Cell. 2014; 159:709–13. https://doi.org/10.1016/j.cell.2014.10.039 [PubMed]
- 91. Blagosklonny MV. Rejuvenating immunity: “anti-aging drug today” eight years later. Oncotarget. 2015; 6:19405–12. https://doi.org/10.18632/oncotarget.3740 [PubMed]
- 92. Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009; 2:ra75. https://doi.org/10.1126/scisignal.2000559 [PubMed]
- 93. Jagannath C, Bakhru P. Rapamycin-induced enhancement of vaccine efficacy in mice. Methods Mol Biol. 2012; 821:295–303. https://doi.org/10.1007/978-1-61779-430-8_18 [PubMed]
- 94. Bravo-San Pedro JM, Senovilla L. Immunostimulatory activity of lifespan-extending agents. Aging (Albany NY). 2013; 5:793–801. https://doi.org/10.18632/aging.100619 [PubMed]
- 95. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 t-cell differentiation. Nature. 2009; 460:108–12. https://doi.org/10.1038/nature08155 [PubMed]
- 96. Ferrer IR, Araki K, Ford ML. Paradoxical aspects of rapamycin immunobiology in transplantation. Am J Transplant. 2011; 11:654–59. https://doi.org/10.1111/j.1600-6143.2011.03473.x [PubMed]
- 97. Turner AP, Shaffer VO, Araki K, Martens C, Turner PL, Gangappa S, Ford ML, Ahmed R, Kirk AD, Larsen CP. Sirolimus enhances the magnitude and quality of viral-specific CD8+ t-cell responses to vaccinia virus vaccination in rhesus macaques. Am J Transplant. 2011; 11:613–18. https://doi.org/10.1111/j.1600-6143.2010.03407.x [PubMed]
- 98. Hurez V, Dao V, Liu A, Pandeswara S, Gelfond J, Sun L, Bergman M, Orihuela CJ, Galvan V, Padrón Á, Drerup J, Liu Y, Hasty P, et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell. 2015; 14:945–56. https://doi.org/10.1111/acel.12380 [PubMed]
- 99. Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB. mTOR inhibition improves immune function in the elderly. Sci Transl Med. 2014; 6:268ra179. https://doi.org/10.1126/scitranslmed.3009892 [PubMed]
- 100. Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, Watkins M, Shavlakadze T, Zhou W, Quinn D, Glass DJ, Klickstein LB. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med. 2018; 10:eaaq1564. https://doi.org/10.1126/scitranslmed.aaq1564 [PubMed]
- 101. Ferrer IR, Wagener ME, Robertson JM, Turner AP, Araki K, Ahmed R, Kirk AD, Larsen CP, Ford ML. Cutting edge: rapamycin augments pathogen-specific but not graft-reactive CD8+ T cell responses. J Immunol. 2010; 185:2004–08. https://doi.org/10.4049/jimmunol.1001176 [PubMed]
- 102. Frasca D, Blomberg BB, Paganelli R. Aging, obesity, and inflammatory age-related diseases. Front Immunol. 2017; 8:1745. https://doi.org/10.3389/fimmu.2017.01745 [PubMed]
- 103. Ventura MT, Casciaro M, Gangemi S, Buquicchio R. Immunosenescence in aging: between immune cells depletion and cytokines up-regulation. Clin Mol Allergy. 2017; 15:21. https://doi.org/10.1186/s12948-017-0077-0 [PubMed]
- 104. Gan B, DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009; 8:1003–06. https://doi.org/10.4161/cc.8.7.8045 [PubMed]
- 105. Blagosklonny MV. Aging, stem cells, and mammalian target of rapamycin: a prospect of pharmacologic rejuvenation of aging stem cells. Rejuvenation Res. 2008; 11:801–08. https://doi.org/10.1089/rej.2008.0722 [PubMed]
- 106. Sousa-Victor P, Perdiguero E, Muñoz-Cánoves P. Geroconversion of aged muscle stem cells under regenerative pressure. Cell Cycle. 2014; 13:3183–90. https://doi.org/10.4161/15384101.2014.965072 [PubMed]
- 107. Ho YH, Méndez-Ferrer S. Microenvironmental contributions to hematopoietic stem cell aging. Haematologica. 2020; 105:38–46. https://doi.org/10.3324/haematol.2018.211334 [PubMed]
- 108. Chen J, Kang JG, Keyvanfar K, Young NS, Hwang PM. Long-term adaptation to hypoxia preserves hematopoietic stem cell function. Exp Hematol. 2016; 44:866–73.e4. https://doi.org/10.1016/j.exphem.2016.04.010 [PubMed]
- 109. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, Suda T. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010; 7:391–402. https://doi.org/10.1016/j.stem.2010.06.020 [PubMed]
- 110. Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci USA. 2012; 109:13314–18. https://doi.org/10.1073/pnas.1205690109 [PubMed]
- 111. Luo Y, Li L, Zou P, Wang J, Shao L, Zhou D, Liu L. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014; 97:20–29. https://doi.org/10.1097/TP.0b013e3182a7fcf8 [PubMed]
- 112. Feng X, Lin Z, Sun W, Hollinger MK, Desierto MJ, Keyvanfar K, Malide D, Muranski P, Chen J, Young NS. Rapamycin is highly effective in murine models of immune-mediated bone marrow failure. Haematologica. 2017; 102:1691–703. https://doi.org/10.3324/haematol.2017.163675 [PubMed]
- 113. Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001; 276:38052–60. https://doi.org/10.1074/jbc.M106703200 [PubMed]
- 114. Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004; 167:399–403. https://doi.org/10.1083/jcb.200408161 [PubMed]
- 115. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124:471–84. https://doi.org/10.1016/j.cell.2006.01.016 [PubMed]
- 116. Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Fürnsinn C, Promintzer M, Anderwald C, Bischof M, Roden M. The mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007; 56:1600–07. https://doi.org/10.2337/db06-1016 [PubMed]
- 117. Leontieva OV, Demidenko ZN, Blagosklonny MV. Rapamycin reverses insulin resistance (IR) in high-glucose medium without causing IR in normoglycemic medium. Cell Death Dis. 2014; 5:e1214. https://doi.org/10.1038/cddis.2014.178 [PubMed]
- 118. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005; 146:1473–81. https://doi.org/10.1210/en.2004-0921 [PubMed]
- 119. Blagosklonny MV. Rapamycin for longevity: opinion article. Aging (Albany NY). 2019; 11:8048–67. https://doi.org/10.18632/aging.102355 [PubMed]
- 120. Taylor R, Al-Mrabeh A, Zhyzhneuskaya S, Peters C, Barnes AC, Aribisala BS, Hollingsworth KG, Mathers JC, Sattar N, Lean ME. Remission of human type 2 diabetes requires decrease in liver and pancreas fat content but is dependent upon capacity for β cell recovery. Cell Metab. 2018; 28:547–56.e3. https://doi.org/10.1016/j.cmet.2018.07.003 [PubMed]
- 121. Steven S, Taylor R. Restoring normoglycaemia by use of a very low calorie diet in long- and short-duration type 2 diabetes. Diabet Med. 2015; 32:1149–55. https://doi.org/10.1111/dme.12722 [PubMed]
- 122. Steven S, Lim EL, Taylor R. Population response to information on reversibility of type 2 diabetes. Diabet Med. 2013; 30:e135–38. https://doi.org/10.1111/dme.12116 [PubMed]
- 123. Nicoll R, Henein MY. Caloric restriction and its effect on blood pressure, heart rate variability and arterial stiffness and dilatation: a review of the evidence. Int J Mol Sci. 2018; 19:751. https://doi.org/10.3390/ijms19030751 [PubMed]
- 124. Blagosklonny MV. Rapamycin and quasi-programmed aging: four years later. Cell Cycle. 2010; 9:1859–62. https://doi.org/10.4161/cc.9.10.11872 [PubMed]
- 125. Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of leigh syndrome. Science. 2013; 342:1524–28. https://doi.org/10.1126/science.1244360 [PubMed]
- 126. Houssaini A, Breau M, Kebe K, Abid S, Marcos E, Lipskaia L, Rideau D, Parpaleix A, Huang J, Amsellem V, Vienney N, Validire P, Maitre B, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 2018; 3:e93203. https://doi.org/10.1172/jci.insight.93203 [PubMed]
- 127. Mitani A, Ito K, Vuppusetty C, Barnes PJ, Mercado N. Restoration of corticosteroid sensitivity in chronic obstructive pulmonary disease by inhibition of mammalian target of rapamycin. Am J Respir Crit Care Med. 2016; 193:143–53. https://doi.org/10.1164/rccm.201503-0593OC [PubMed]
- 128. McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, Brown KK, Lynch JP
3rd , Goldberg HJ, et al, National Institutes of Health Rare Lung Diseases Consortium, and MILES Trial Group. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011; 364:1595–606. https://doi.org/10.1056/NEJMoa1100391 [PubMed] - 129. Blagosklonny MV. Fasting and rapamycin: diabetes versus benevolent glucose intolerance. Cell Death Dis. 2019; 10:607. https://doi.org/10.1038/s41419-019-1822-8 [PubMed]
- 130. Pavlakis M, Goldfarb-Rumyantzev AS. Diabetes after transplantation and sirolimus: what’s the connection? J Am Soc Nephrol. 2008; 19:1255–56. https://doi.org/10.1681/ASN.2008050474 [PubMed]
- 131. Veroux M, Tallarita T, Corona D, Sinagra N, Giaquinta A, Zerbo D, Guerrieri C, D’Assoro A, Cimino S, Veroux P. Conversion to sirolimus therapy in kidney transplant recipients with new onset diabetes mellitus after transplantation. Clin Dev Immunol. 2013; 2013:496974. https://doi.org/10.1155/2013/496974 [PubMed]
- 132. Tanimura J, Nakagawa H, Tanaka T, Kikuchi A, Osada S, Tanaka Y, Tokuyama K, Takamura T. The clinical course and potential underlying mechanisms of everolimus-induced hyperglycemia. Endocr J. 2019; 66:615–20. https://doi.org/10.1507/endocrj.EJ18-0542 [PubMed]
- 133. Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020; 6:14. https://doi.org/10.1038/s41421-020-0153-3 [PubMed]
- 134. Omarjee L, Janin A, Perrot F, Laviolle B, Meilhac O, Mahe G. Targeting t-cell senescence and cytokine storm with rapamycin to prevent severe progression in COVID-19. Clin Immunol. 2020; 216:108464. https://doi.org/10.1016/j.clim.2020.108464 [PubMed]
- 135. Zheng Y, Li R, Liu S. Immunoregulation with mTOR inhibitors to prevent COVID-19 severity: a novel intervention strategy beyond vaccines and specific antiviral medicines. J Med Virol. 2020; 10:1002. https://doi.org/10.1002/jmv.26009 [PubMed]
- 136. Maiese K. The mechanistic target of rapamycin (mTOR): novel considerations as an antiviral treatment. Curr Neurovasc Res. 2020. [Epub ahead of print]. https://doi.org/10.2174/1567202617666200425205122 [PubMed]
- 137. Lehrer S. Inhaled biguanides and mTOR inhibition for influenza and coronavirus (review). World Acad Sci J. 2020; 2:1. https://doi.org/10.3892/wasj.2020.42 [PubMed]
- 138. Zou J, Zou P, Wang J, Li L, Wang Y, Zhou D, Liu L. Inhibition of p38 MAPK activity promotes ex vivo expansion of human cord blood hematopoietic stem cells. Ann Hematol. 2012; 91:813–23. https://doi.org/10.1007/s00277-011-1397-7 [PubMed]
- 139. Ranadheera C, Coombs KM, Kobasa D. Comprehending a killer: the Akt/mTOR signaling pathways are temporally high-jacked by the highly pathogenic 1918 influenza virus. EBioMedicine. 2018; 32:142–63. https://doi.org/10.1016/j.ebiom.2018.05.027 [PubMed]
- 140. Kindrachuk J, Ork B, Hart BJ, Mazur S, Holbrook MR, Frieman MB, Traynor D, Johnson RF, Dyall J, Kuhn JH, Olinger GG, Hensley LE, Jahrling PB. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for middle east respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother. 2015; 59:1088–99. https://doi.org/10.1128/AAC.03659-14 [PubMed]
- 141. Seminari E, Colaneri M, Sambo M, Gallazzi I, Di Matteo A, Roda S, Bruno R, and COVID19 IRCCS San Matteo Pavia Task Force. SARS cov-2 infection in a renal-transplanted patient: a case report. Am J Transplant. 2020. [Epub ahead of print]. https://doi.org/10.1111/ajt.15902 [PubMed]
- 142. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, and HLH Across Speciality Collaboration, UK. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020; 395:1033–34. https://doi.org/10.1016/S0140-6736(20)30628-0 [PubMed]
- 143. Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M. The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020; 53:25–32. https://doi.org/10.1016/j.cytogfr.2020.05.003 [PubMed]
- 144. Rolt A, Nair A, Cox LS. Optimisation of a screening platform for determining IL-6 inflammatory signalling in the senescence-associated secretory phenotype (SASP). Biogerontology. 2019; 20:359–71. https://doi.org/10.1007/s10522-019-09796-4 [PubMed]
- 145. Chiang PH, Wang L, Bonham CA, Liang X, Fung JJ, Lu L, Qian S. Mechanistic insights into impaired dendritic cell function by rapamycin: inhibition of Jak2/Stat4 signaling pathway. J Immunol. 2004; 172:1355–63. https://doi.org/10.4049/jimmunol.172.3.1355 [PubMed]
- 146. Wang CH, Chung FT, Lin SM, Huang SY, Chou CL, Lee KY, Lin TY, Kuo HP. Adjuvant treatment with a mammalian target of rapamycin inhibitor, sirolimus, and steroids improves outcomes in patients with severe H1N1 pneumonia and acute respiratory failure. Crit Care Med. 2014; 42:313–21. https://doi.org/10.1097/CCM.0b013e3182a2727d [PubMed]