



Introduction
Cancer in the brain is significantly less common than in other organs, but disproportionately contributes to higher rates of mortality [1, 2]. Although prognostic factors have been identified (e.g. MGMT promoter methylation), long-term treatments remain ineffective, in part due to diverse challenges in research and development as well as an incomplete understanding of brain tumor biology [3–5]. Treatment options for central nervous system (CNS) tumors are limited (e.g. chemotherapy, radiotherapy (RT), surgical resection etc.) and overall mortality rates exceed 60% within 5 years of diagnosis [6].
In the United States, the prevalence of brain tumors is highest in the 50-59 age group, followed by the 60-69 and 70-79 cohorts, respectively [7]. While the frequency of older individuals diagnosed with these insidious neuropathologies continues to increase, advancing age itself is associated with poorer prognosis [8–12]. For instance, elderly patients maintain elevated mortality rates compared to younger individuals and benefit from shorter courses of RT because of increased susceptibility to side effects of full-dose RT [12, 13]. Given their vulnerability, as well as the contemporary increase in aging demographics, determining the underlying biological mechanisms of tumor pathology in the aging brain and improving treatment options has never been more urgent [14]. Since metabolic syndrome (e.g. hyperglycemia) is an independent risk factor for worse prognosis in patients with certain CNS tumors, bioenergetic processes in particular are emerging as important variables in tumor neuropathology [15].
Among primary cell types implicated in tumor pathogenesis, minimal contributions are observed from non-glia; less than 10% of all tumors manifest as lymphomas, meningiomas, embryonal tumors, or choroid plexus carcinomas, among others [6]. Although neuron-specific oncogenesis has been documented (e.g. neurocytoma, ganglioneurocytoma), its prevalence is extremely rare (~1%) and is routinely characterized by mixed neuronal-glial presentation [16]. Additionally, while microglia are sufficient to augment tumor progression in the CNS, microglia-specific cancers appear to be absent [17, 18]. Conversely, more than 75% of cancers in the CNS are of glial origin [6]. These tumors can be classified and graded in severity utilizing an array of molecular markers and assessment of specific genetic mutations (e.g. isocitrate dehydrogenase; IDH) [19]. Within glioma subtypes, glioblastomas account for more than half of all diagnoses and are the most lethal, with a 5-year survival rate of less than 6% [20]. As tumors of glial origin account for the vast majority of all CNS cancers (i.e. pilocytic astrocytoma, oligodendroglioma, diffuse astrocytoma, anaplastic astrocytoma, glioblastoma), age-dependent variation in glial functioning may particularly contribute to tumor pathology in the brain [6].
The unique role of glia in modulating CNS bioenergetics is often underappreciated and is reminiscent of the metabolic characteristics found in cancer cells of other organs. Here, glia can preferentially process glucose to produce lactate, which is then exported to serve as a primary energy source for energy-demanding neuronal populations [21, 22]. Such predominant production of lactate and its subsequent extrusion to neurons, rather than glucose utilization in mitochondrial oxidative phosphorylation, is indeed similar to the metabolic characteristics commonly observed in cancer cells in other organs (i.e. Warburg effect; aerobic glycolysis) [23, 24]. Within the CNS, astrocytes in particular have long been associated with metabolic homeostasis and are the most common cell type associated with tumor pathology [6, 25, 26]. This suggests the unique metabolic processes and related pathways in these glia may contribute to glioma pathology in the aging CNS as well as poorer outcomes in aged patients.
Along with a continual production of lactate, cancer cells often maintain hyperactivation of the PAM (PI3K/AKT/mTOR) pathway [27, 28]. This evolutionary conserved metabolic pathway can be governed by upstream insulin signaling and may be one of the most diverse in mammals; it affects a range of cellular molecules and functions, from nutrient transporters and metabolic enzymes to gene expression and autophagy, respectively [29, 30]. Interestingly, variation in bioenergetic demands, insulin signaling (e.g. insulin resistance) and downstream PAM signaling (e.g. mTOR dysregulation) are characteristic of the aged brain and are associated with age-related neuropathogenesis [31–34]. Given that glia are responsible for maintaining energy homeostasis of the CNS by utilizing similar metabolic pathways of cancer cells (i.e. aerobic glycolysis), chronic alterations in bioenergetic demands during aging may aberrantly regulate PAM-dependent mechanisms in glia (e.g. cell cycle progression, autophagy, neuroinflammation); in turn, this could contribute to tumor pathology in the CNS and facilitate poorer prognosis in the elderly.
Cancer bioenergetics and PAM signaling
The metabolism of cancer cells is distinct and the implications of these bioenergetic characteristics are increasingly relevant in assessing the etiology of oncogenesis as well as tumor development [35, 36]. For instance, dietary high-fructose corn syrup was recently shown to directly contribute to tumor formation in murine intestines [37]. Similar to glia in the brain (see below), glucose in cancer cells is predominantly converted to lactate via glycolysis, rather than being fully oxidized via respiration in mitochondria. The increased glucose flux and conversion to lactate despite the presence of adequate oxygen levels (i.e. aerobic glycolysis) as well as functional mitochondria has been termed the “Warburg Effect” [24, 38, 39]. While this produces a higher rate yet lower yield of ATP production, it is also thought to generate substrates that facilitate anabolic processes during cell proliferation (e.g. lipids, proteins, nucleotides), promote intercellular oncogenic signaling (e.g. reactive oxygen species; ROS) and disrupt functioning in surrounding non-oncogenic cells [39–42]. Coupled with altered vasculature as well as a continuous supply of glucose and glycolysis-promoting factors (e.g. insulin), such distinct metabolism of cancer cells promotes their fitness at the expense of their non-oncogenic neighbors [43, 44].
In addition to metabolic reprogramming, cancer cells routinely display hyperactivation of Phosphoinositide 3-kinase (PI3K)-dependent pathways, a signaling cascade that controls multiple steps in cellular bioenergetics and can promote aerobic glycolysis [27, 45]. PI3K, and its downstream effectors AKT/mTOR (PAM), constitute an evolutionarily conserved metabolic pathway that can be modulated by an array of factors, including insulin signaling [46]. In brief, insulin or other growth factors (e.g. insulin-like growth factors; IGF) bind to requisite receptors on the cell surface, causing insulin-receptor substrate phosphorylation, downstream PI3K/AKT/mTOR activation and ultimately increased glucose uptake [28, 47]. Consequently, hyperinsulinemia induced PAM activation can promote tumor progression and is associated with increased mortality rates in non-CNS cancers [48–52]. Conversely, the mitigation of insulin levels in non-CNS cancer cells can inhibit oncogenesis, while insulin-induced proliferation in these cells can be reversed by inhibiting PAM activation [53, 54].
Given its diverse roles in maintaining cellular bioenergetics, aberrant PAM signaling in cancer is associated with variation across several interdependent homeostatic mechanisms, including cell cycle progression, autophagy and inflammation. For instance, activation of PI3K and subsequent phosphorylation of AKT is necessary for mitigating the inhibitory regulation of cyclin-dependent kinases in cellular proliferation, thus promoting cell cycle entry in cancer cells [55, 56]. As a potent autophagy inhibitor, mTOR1 is also a notable downstream target of PI3K activation in cancer [57, 58]. Specifically, mTOR1 can suppress a variety of rate limiting steps in this protein degradation system, including the inhibition of autophagosome dynamics (e.g. nucleation, elongation, termination) and the dysregulation of transcription factors responsible for the expression of autophagy-dependent genes (e.g. TFEB) [59, 60]. Additionally, PAM activation can dysregulate host immune mechanisms and promote tumor progression, while the pharmacological inhibition of PI3K can mitigate these processes [61–63]. For instance, PI3K activation inhibits anti-cancer capacities in T-cells to promote tumorigenesis, and PI3K inhibition can mitigate the recruitment of myeloid cells to the tumor microenvironment that otherwise promote tumor growth [64–66]. Thus, the metabolic demands of oncogenic cells and associated hyperactivation of PAM signaling may potentiate pathogenesis through these altered homeostatic mechanisms.
Glia bioenergetics and PAM signaling
The energy demands of the CNS necessitate efficient regulation and are uniquely dependent on glia. Indeed, the brain requires more energy compared to other organs (i.e. 2% of body mass but 20% of total body energy), the demands between cells are disproportionate (i.e. 80-90% of all energy in the brain is used by neurons for action potential generation, maintaining resting membrane potential etc.) and its lack of reserves (e.g. limited adipose tissue) must be compensated by continuous access to essential macronutrients [67–70]. Although neurons themselves have been shown to process glucose for energy, efficient bioenergetics in the CNS involve glucose conversion to lactate by glia, which is then extruded to serve as a principal energy source for neuronal populations [21, 22].
In a process termed the astrocyte-to-neuron lactate shuttle (ANLS) hypothesis, astrocyte pericapillary end-feet initially take up glucose from the resource-rich vasculature in conjunction with pericytes, convert it to lactate via lactate dehydrogenase (LDH) and transport the lactate via monocarboxylate transporters (MCT) to neurons, where it serves as an energy substrate for ATP synthesis in neuronal mitochondria [23, 71]. Such metabolic processes are linked to activity dependent neurotransmission, where evoked release of vesicular glutamate at distal axonal terminals is taken up by astrocytes, converted to glutamine and transported back to neurons, which are otherwise incapable of de novo synthesis of such neurotransmitter precursors [72–74]. Along with fluctuations in metabolic demands, neuronal activity may directly contribute to tumor pathogenesis via glutamate signaling and other small molecules produced by neuronal firing (e.g. neuroligin-3) [75, 76]. As tumor growth induced by neuronal activity coincides with cancer cell colonization at synaptic junctions (i.e. where astrocytes would otherwise be located given the ANLS), further investigations are encouraged to determine how differing functioning of glia during aging may alter this interaction with neurons to facilitate tumor development [77].
Although direct evidence for some of these mechanisms is lacking, and other processes undoubtedly contribute to energy homeostasis in the CNS (e.g. neuronal oxidative phosphorylation and lactate export, pentose phosphate shunt, glycogen turnover, acetate use etc.), accumulating evidence implicates the importance of glycolysis in astrocytes for maintaining CNS bioenergetics [21, 78–80]. More recently, similar lactate-dependent bioenergetic processes have been extended to oligodendrocytes, the second most common cell type associated with CNS cancers [81–83]. It should be noted that precursors of both astrocytes -and oligodendrocytes may serve as progenitors for tumorigenesis [84, 85]. Given that astrocytic support for oligodendrocytes via extracellular vesicles is compromised in aging, future experiments should examine if age-related metabolic variations influence the interplay between astrocytes and oligodendrocytes to promote pathological processes in CNS cancers [86].
While the metabolic demands of glia are unique in the CNS and emulate cancer metabolism (e.g. glycolysis instead of respiration), variation in PAM signaling can also accompany these metabolic variations in the absence of overt pathogenesis. In astrocytes, enhanced lactate production triggered by oxidative stress is dependent on PAM activation [87]. Similarly, insulin stimulation of astrocytes results in a dose-dependent activation of downstream PI3K-dependent effectors, including AKT and GSK3 [88]. Although evidence of differing glycolysis associated with PAM variation in non-oncogenic oligodendrocytes is lacking, increased metabolic demands in these cells (e.g. myelination) are indeed associated with increased PI3K-dependent signaling [89, 90]. Moreover, metabolic demands of astrocytes and ensuing activation of PAM signaling can precipitate similar signaling cascades in their oligodendrocyte counterparts [91].
Elevated glucose uptake, increased glycolysis and altered metabolic pathways are well characterized features of glioma biology, similar to other cancer types [92, 93]. Such elevated glycolysis is most commonly associated with genetic dysregulation of PAM pathways, which are evident in over 85% of glioblastoma cases and can predict prognosis in patients [94, 95]. While aberrant PAM activation accompanies oncogenesis across various glioma subtypes, the induction of PAM signaling in vitro is sufficient to promote cellular proliferation in human glioblastoma as well as oligodendroglioma cells [96–100]. Experiments utilizing similar cell culture systems suggest aerobic glycolysis and sufficient lactate production in brain tumors is inherently dependent on PAM pathway induction, while pharmacological inhibition of this signaling pathway in the brains of mice can mitigate the growth of orthotopic tumors [101, 102]. Evidence also suggests the involvement of PAM is conserved across tumors of varying glial origin, provided that in vivo inducible expression of PI3K is capable of precipitating oligodendroglioma oncogenesis, while deletion of PI3K-inhibitory factor (PTEN) can facilitate substantially more aggressive tumor progression [103].
Similar to other cancers, dysregulated PAM signaling in glioma cells is associated with variation across several important homeostatic mechanisms, such as cell cycle progression, autophagy and inflammation. In glioblastoma cell lines (i.e. U87MG and U251MG), and ex vivo glioma brain slices, activation of PAM signaling is necessary for cell cycle progression and tumor expansion, potentially due to PI3K/AKT-suppression of cyclin D1 [98, 104]. Additionally, glioma cells require PAM-induced suppression of autophagy to maintain viability, while suppressed autophagy and tumor infiltration in orthotopic glioma models is dependent on PAM activation [105, 106]. Glioma similarly activates PAM pathways to modulate cytokine signaling and recruit proinflammatory microglia to the microenvironment, which promote tumor growth [107–111]. Given the unique metabolic demands of glia (i.e. aerobic glycolysis) and the dysregulation of PAM signaling observed in glioma cells, variation in CNS bioenergetics during aging and associated PAM signaling in glia may contribute to pathological processes in CNS cancers.
Potentiation of glial PAM in aging: potential mechanisms in brain tumors
Due to their role in maintaining energy homeostasis in the brain, alterations in CNS bioenergetics that accompany aging may modulate the metabolic demands of glia and dysregulate their PAM signaling, thus contributing to tumor pathology in elderly patients. Dysregulation of insulin and glucose processing in the CNS is characteristic of aging as well as age-related neurodegenerative diseases (e.g. Alzheimer’s Disease, AD; Parkinson’s Disease, PD) [112–115]. Such deficiencies in energy dynamics during aging are due to several factors, including impairments in mitochondrial oxidative phosphorylation, altered expression of glucose transporters, dysregulated redox homeostasis, and adaptations to insulin signaling itself [116–119]. Furthermore, a limited nutrient supply and hypoperfusion may exacerbate the disproportional strains on aerobic glycolysis in aging [120, 121]. While deficits in energy utilization are characteristic of the aging CNS, astrocytes can compensate for metabolic deficiencies in neurons (e.g. increase glucose uptake, upregulate glycolysis) [122–124]. Interestingly, variation in these bioenergetic demands can trigger a direct upregulation in glial glycolysis, sometimes referred to as the “Inverse Warburg Effect” [125]. Indeed, astrocytes can better cope with hyperglycemic fluctuations compared to neurons, and they can directly alter their metabolism as well as PAM signaling in response to insulin stimulation [88, 123, 126].
Given the role of glia, specifically astrocytes, in maintaining metabolic homeostasis in the CNS, variation in such metabolic demands during aging may aberrantly regulate PAM signaling in these cells to promote tumor pathology. Indeed, elevations of insulin can trigger PAM signaling in astrocytes, while the induction of aerobic glycolysis in astrocytes is itself dependent on PAM activation [87, 88]. However, astrocytic adaptations to these conditions may be maladaptive. For instance, disrupted insulin signaling in astrocytes following metabolic fluctuations may inhibit the transport of insulin/glucose into the brain [112, 127]. In addition, cell cycle progression in astrocytes can be modulated by hyperglycemic conditions, while proliferation and survival of CNS cancer cells is inherently coupled to insulin-dependent PAM signaling [100, 128–130]. Furthermore, accumulating evidence indicates certain subpopulations of astrocytes with distinct metabolic characteristics are more prone to oncogenesis, while such glial heterogeneity significantly contributes to tumor development [131, 132]. Conversely, the reduction in glycolysis at the expense of increased oxidative phosphorylation (i.e. “Anti-Warburg Effect”) is sufficient to trigger glioma differentiation into non-oncogenic astrocytes [133]. Moreover, whereas the induction of PAM signaling is observed in glial pathogenesis and tumor development, its mitigation is associated with extended longevity and CNS homeostasis in aging [31, 134, 135]. Therefore, metabolic inefficiencies in the brain during aging may converge on glial bioenergetic demands to dysregulate PAM-dependent mechanisms, promote tumor pathology, and contribute to poor prognosis in elderly patients (Figure 1).
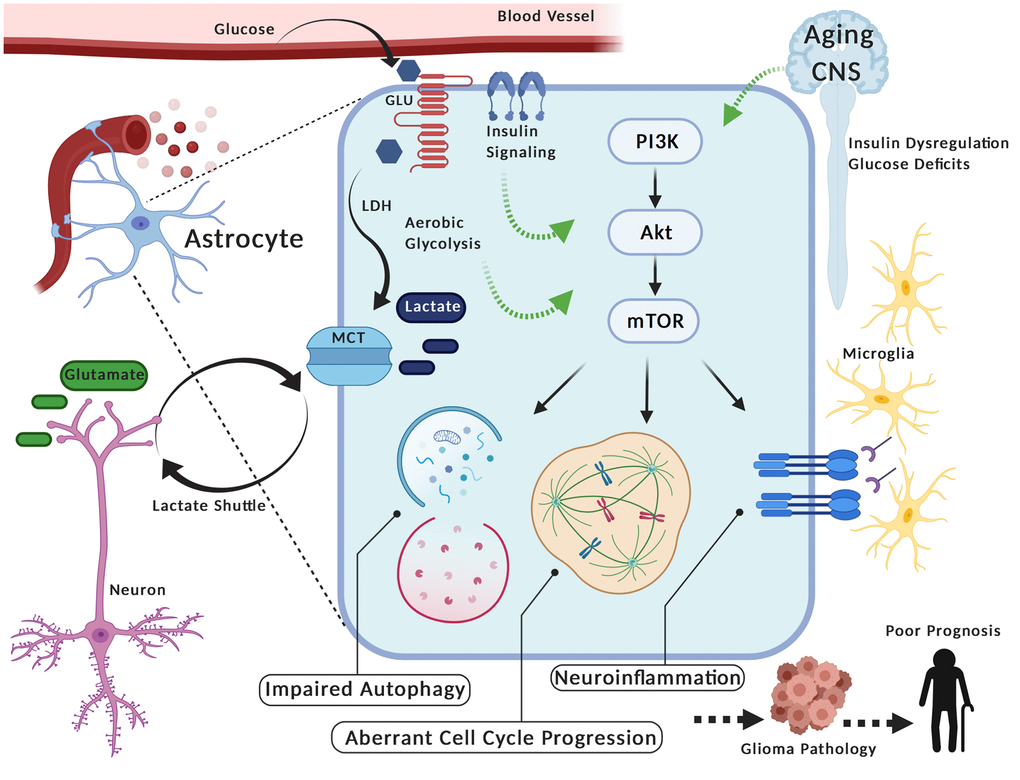
Figure 1. PAM (PIK3/AKT/mTOR) Signaling in glia during aging may contribute to glioma pathology. Astrocytes take up glucose, convert it to lactate (i.e. aerobic glycolysis) via lactate dehydrogenase (LDH) and transport it via monocarboxylate transporters (MCT) to neurons (i.e. Lactate Shuttle) where it serves as an energy substrate. These glia can alter their metabolism and also re-uptake glutamate in response to neuronal firing activity. Interestingly, the aerobic glycolysis in astrocytes is reminiscent of energy production in cancer cells, including gliomas. Furthermore, variation in metabolic activity and/or insulin stimulation in astrocytes corresponds with activation of PAM (PIK3/AKT/mTOR), a pathway linked to oncogenic processes in tumor cells (e.g. aberrant cell cycle progression, impaired autophagy, neuroinflammation). Thus, metabolic fluctuations in the central nervous system (CNS) during aging (e.g. insulin dysregulation, deficits in glucose utilization) may aberrantly potentiate PAM signaling in these glia, given their prominent role in metabolic homeostasis. In turn, this could contribute to pathogenic processes of gliomas and facilitate poor prognosis in elderly individuals. Created with Biorender.com.
Similar to observations in gliomas, dysregulation of PAM-dependent mechanisms is observed in age-related neurodegenerative conditions, suggesting their contributions to CNS disease progression in aging. Indeed, PAM induction of aberrant cell cycle progression, impaired autophagy and neuroinflammation is implicated in AD pathogenesis [136–138]. Additionally, data suggest that patients displaying AD symptomology maintain poorer cancer-related outcomes, including diagnosis with more advanced stages of cancer, decreased life expectancies and increased mortality rates [139–143]. Such activation of PAM-dependent mechanisms and downstream oncogenesis is seemingly contradicted by population-based cohort studies, which indicate AD patients maintain reduced risk of cancer by up to ~50% and cancer patients maintain reduced risk of AD by up to ~35% [144, 145]. However, it is important to note this correlation may be confounded by sampling errors in data collection (i.e. demented patients are less likely to be screened for cancer and cancer patients may not live long enough to develop dementia) [146, 147]. Therefore, PAM signaling may nonetheless represent an important node of CNS homeostasis in aging, whereby its dysregulation can facilitate impairments in crucial mechanisms that promote disease progression in the brain.
In addition to PAM, age-related variation in several other glial mechanisms may influence glioma pathology and contribute to poor prognosis among elderly patients. For instance, aging astrocytes can develop distinct expression profiles, characterized by upregulation of multiple immune pathways (e.g. complement system, antigen presentation), increased immune cell attractants (e.g. CXCL5, CXCL10) and elevations in proinflammatory cytokines [148–150]. Given that variation in neuroinflammatory microenvironments can augment tumor progression, the inflammatory profiles of aged astrocytes may facilitate glioma pathology and influence cancer-related outcomes in aged individuals [151, 152]. Although balanced ROS levels are necessary for optimal functioning, astrocytes from the aging brain also display increased levels of oxidative stress compared to their youthful counterparts, which may exacerbate the elevated levels of reactive oxygen species otherwise induced by metabolically demanding glioma cells; this suggests the mitigation of oxidative stress in aging glia may serve as a novel target for treatment of glioma in the elderly [153, 154].
Conclusions
The present mechanistic model suggests variation in metabolic demands and aberrant PAM signaling in glia during aging, as well as its downstream consequences (i.e. cell cycle progression, impaired autophagy and neuroinflammation), are contributing factors to glioma pathology in elderly patients. Current postulations argue that tumor development may be attributed to aberrantly self-renewing, stem-like cells found within gliomas, or hypermutations in oncogenic pathways [155–157]. The latter theory is supported by observations of deficient DNA repair mechanisms in some malignancies, as well as the targeting of DNA repair mechanisms in commonly used glioma chemotherapy (e.g. TMZ) [158, 159]. Interestingly, recent evidence suggests the characteristic genetic diversity of varying glioma pathologies is likely due to culminated transcriptomic and epigenomic modifications gained from differing growth environments [160, 161]. Thus, by suggesting the unique growth environment and bioenergetic demands of glia in the aged CNS potentiate tumor pathology via PAM signaling, our model complements existing theories of glioma etiology.
The emphasis on host cell metabolic demands in aging as an important factor in glioma development supports prior data which suggests these demands can interact with viral oncoproteins to promote tumorigenesis [162]. Although neurotropic viruses encode protein antagonists of innate immune mechanisms to evade detection of viral RNA or DNA, these can inadvertently inhibit tumor suppressors and facilitate tumor progression [163–165]. For instance, p53 can be targeted by protein products in multiple viruses, including polyomaviruses (e.g. JCV), retroviruses (e.g. HIV, SIV) and herpesviruses (e.g. CMV) [166–169]. As one of the most commonly detected viruses in CNS tumors, the JCV encoded T-antigen has been shown to inactivate p53’s tumor suppressing functions and promote cell cycle progression, while these capacities of T-antigen can be independently regulated by glucose availability or insulin signaling cascades [170–173]. Furthermore, this inactivation of tumor suppressors by neurotropic viruses, in addition to their capacity to induce aerobic glycolysis and dysregulate PAM signaling, may interact with metabolic demands in aging glia to promote tumor development in the brain [174, 175].
Our model maintains several limitations. First, the role of glia themselves in regulating CNS bioenergetics remains actively debated, with some arguing that fluctuations in neuronal metabolism (e.g. glucose uptake, oxidative phosphorylation, lactate export etc.), rather than variations in astrocytic glycolysis, are predominantly responsible for energy homeostasis in the brain [80]. Additionally, while the current review was intended to highlight the unique position of glia as a potential intermediary between PAM signaling and age-related metabolic demands, this parsimonious framework does not incorporate the involvement or crosstalk with other potential pathways in glioma, particularly the MAPK/ERK cascade [176, 177]. Third, while this model takes into account the molecular signatures of glioma in patients, as well as preclinical gain/loss of function experiments, the limited empirical support for the proposed framework is underscored by the broader discordance between neurobiological and cancer research, which hinders the assessment of causal molecular mechanisms [3].
Although limitations persist, the modulation of PAM signaling in glia during aging may elucidate several therapeutic opportunities for brain tumor treatment, including the beneficial role of non-pharmacological interventions. In particular, the inhibition of PI3K’s downstream effectors, AKT and mTOR, may prove to reliably mitigate proliferation, motility and viability in glioma cells [178–180]. Additionally, PI3K inhibitors are emerging as potent antagonistic agents in various types of cancer, including gliomas [102, 181, 182]. While cancer cells have been shown to overcome this inhibition, evidence from preclinical models suggest the combination of dietary manipulations as well as PI3K inhibitors can augment the inhibition of PAM signaling and increase therapeutic efficacy [28]. Compared to mice treated with PI3K inhibitors alone, mice maintained on a ketogenic diet during treatment display significantly decreased mortality rates following intracranial implantation of aggressive human gliomas [183]. Indeed, effective regulation of insulin levels via a balanced diet or aerobic exercise is a recognized strategy to prevent age-related risks for cancer as well as neurodegenerative diseases [184–186]. Although further validation of these targets is required, the modulation of PAM signaling to mitigate glioma progression in the aging CNS may prove efficacious and merits consideration.
Author Contributions
Conceived concept for manuscript: MD, MW and KK; Conducted literature search: MD; Wrote manuscript: MD, MW and KK.
Acknowledgments
The authors wish to thank past and present members of the Department of Neuroscience and Center for Neurovirology for their support, as well as the sharing of ideas and reagents.
Conflicts of Interest
No potential conflicts of interest were reported by the authors.
Funding
This work was made possible by grant T32MH079785 and grants awarded by NIH to KK.
References
- 1. Bondy ML, Scheurer ME, Malmer B, Barnholtz-Sloan JS, Davis FG, Il’yasova D, Kruchko C, McCarthy BJ, Rajaraman P, Schwartzbaum JA, Sadetzki S, Schlehofer B, Tihan T, et al, and Brain Tumor Epidemiology Consortium. Brain tumor epidemiology: consensus from the brain tumor epidemiology consortium. Cancer. 2008; 113:1953–68. https://doi.org/10.1002/cncr.23741 [PubMed]
- 2. Chien LN, Gittleman H, Ostrom QT, Hung KS, Sloan AE, Hsieh YC, Kruchko C, Rogers LR, Wang YF, Chiou HY, Barnholtz-Sloan JS. Comparative brain and central nervous system tumor incidence and survival between the United States and Taiwan based on population-based registry. Front Public Health. 2016; 4:151. https://doi.org/10.3389/fpubh.2016.00151 [PubMed]
- 3. Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, Gottardo N, Gutmann DH, Hargrave D, Holland EC, Jones DT, Joyce JA, Kearns P, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol. 2019; 16:509–20. https://doi.org/10.1038/s41571-019-0177-5 [PubMed]
- 4. Butler M, Pongor L, Su YT, Xi L, Raffeld M, Quezado M, Trepel J, Aldape K, Pommier Y, Wu J. MGMT status as a clinical biomarker in glioblastoma. Trends Cancer. 2020; 6:380–91. https://doi.org/10.1016/j.trecan.2020.02.010 [PubMed]
- 5. Gorlia T, van den Bent MJ, Hegi ME, Mirimanoff RO, Weller M, Cairncross JG, Eisenhauer E, Belanger K, Brandes AA, Allgeier A, Lacombe D, Stupp R. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol. 2008; 9:29–38. https://doi.org/10.1016/S1470-2045(07)70384-4 [PubMed]
- 6. Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017; 19:v1–88. https://doi.org/10.1093/neuonc/nox158 [PubMed]
- 7. Howlader N, Noone A, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis D, Chen H, Feuer E, Cronin K. SEER Cancer Statistics Review, 1975-2017. National Cancer Institute. Bethesda, MD. 2019.
- 8. Greig NH, Ries LG, Yancik R, Rapoport SI. Increasing annual incidence of primary Malignant brain tumors in the elderly. J Natl Cancer Inst. 1990; 82:1621–24. https://doi.org/10.1093/jnci/82.20.1621 [PubMed]
- 9. Hess KR, Broglio KR, Bondy ML. Adult glioma incidence trends in the United States, 1977-2000. Cancer. 2004; 101:2293–99. https://doi.org/10.1002/cncr.20621 [PubMed]
- 10. Young JS, Chmura SJ, Wainwright DA, Yamini B, Peters KB, Lukas RV. Management of glioblastoma in elderly patients. J Neurol Sci. 2017; 380:250–55. https://doi.org/10.1016/j.jns.2017.07.048 [PubMed]
- 11. Kita D, Ciernik IF, Vaccarella S, Franceschi S, Kleihues P, Lütolf UM, Ohgaki H. Age as a predictive factor in glioblastomas: population-based study. Neuroepidemiology. 2009; 33:17–22. https://doi.org/10.1159/000210017 [PubMed]
- 12. Laperriere N, Weller M, Stupp R, Perry JR, Brandes AA, Wick W, van den Bent MJ. Optimal management of elderly patients with glioblastoma. Cancer Treat Rev. 2013; 39:350–57. https://doi.org/10.1016/j.ctrv.2012.05.008 [PubMed]
- 13. Malmström A, Grønberg BH, Marosi C, Stupp R, Frappaz D, Schultz H, Abacioglu U, Tavelin B, Lhermitte B, Hegi ME, Rosell J, Henriksson R, and Nordic Clinical Brain Tumour Study Group (NCBTSG). Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012; 13:916–26. https://doi.org/10.1016/S1470-2045(12)70265-6 [PubMed]
- 14. Organization WH. (2015). World report on ageing and health. (Luxembourg: World Health Organization).
- 15. Rogers LR, Ostrom QT, Schroer J, Vengoechea J, Li L, Gerson S, Nock CJ, Machtay M, Selman W, Lo S, Sloan AE, Barnholtz-Sloan JS. Association of metabolic syndrome with glioblastoma: a retrospective cohort study and review. Neurooncol Pract. 2020; 7:541–48. https://doi.org/10.1093/nop/npaa011 [PubMed]
- 16. Shin JH, Lee HK, Khang SK, Kim DW, Jeong AK, Ahn KJ, Choi CG, Suh DC. Neuronal tumors of the central nervous system: radiologic findings and pathologic correlation. Radiographics. 2002; 22:1177–89. https://doi.org/10.1148/radiographics.22.5.g02se051177 [PubMed]
- 17. Gutmann DH, Kettenmann H. Microglia/brain macrophages as central drivers of brain tumor pathobiology. Neuron. 2019; 104:442–49. https://doi.org/10.1016/j.neuron.2019.08.028 [PubMed]
- 18. Soto MS, Sibson NR. The multifarious role of microglia in brain metastasis. Front Cell Neurosci. 2018; 12:414. https://doi.org/10.3389/fncel.2018.00414 [PubMed]
- 19. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016; 131:803–20. https://doi.org/10.1007/s00401-016-1545-1 [PubMed]
- 20. Silantyev AS, Falzone L, Libra M, Gurina OI, Kardashova KS, Nikolouzakis TK, Nosyrev AE, Sutton CW, Mitsias PD, Tsatsakis A. Current and future trends on diagnosis and prognosis of glioblastoma: from molecular biology to proteomics. Cells. 2019; 8:863. https://doi.org/10.3390/cells8080863 [PubMed]
- 21. Dienel GA. Brain glucose metabolism: integration of energetics with function. Physiol Rev. 2019; 99:949–1045. https://doi.org/10.1152/physrev.00062.2017 [PubMed]
- 22. Jha MK, Morrison BM. Glia-neuron energy metabolism in health and diseases: new insights into the role of nervous system metabolic transporters. Exp Neurol. 2018; 309:23–31. https://doi.org/10.1016/j.expneurol.2018.07.009 [PubMed]
- 23. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. 1994; 91:10625–29. https://doi.org/10.1073/pnas.91.22.10625 [PubMed]
- 24. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016; 41:211–18. https://doi.org/10.1016/j.tibs.2015.12.001 [PubMed]
- 25. Santello M, Toni N, Volterra A. Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci. 2019; 22:154–66. https://doi.org/10.1038/s41593-018-0325-8 [PubMed]
- 26. O’Brien ER, Howarth C, Sibson NR. The role of astrocytes in CNS tumors: pre-clinical models and novel imaging approaches. Front Cell Neurosci. 2013; 7:40. https://doi.org/10.3389/fncel.2013.00040 [PubMed]
- 27. Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020; 20:74–88. https://doi.org/10.1038/s41568-019-0216-7 [PubMed]
- 28. Hopkins BD, Goncalves MD, Cantley LC. insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat Rev Endocrinol. 2020; 16:276–83. https://doi.org/10.1038/s41574-020-0329-9 [PubMed]
- 29. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014; 4:64. https://doi.org/10.3389/fonc.2014.00064 [PubMed]
- 30. Xu F, Na L, Li Y, Chen L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020; 10:54. https://doi.org/10.1186/s13578-020-00416-0 [PubMed]
- 31. Heras-Sandoval D, Avila-Muñoz E, Arias C. The Phosphatidylinositol 3-Kinase/mTor Pathway as a Therapeutic Target for Brain Aging and Neurodegeneration. Pharmaceuticals (Basel). 2011; 4:1070–87. https://doi.org/10.3390/ph4081070
- 32. Gabbouj S, Ryhänen S, Marttinen M, Wittrahm R, Takalo M, Kemppainen S, Martiskainen H, Tanila H, Haapasalo A, Hiltunen M, Natunen T. Altered insulin signaling in Alzheimer’s disease brain - special emphasis on PI3K-Akt pathway. Front Neurosci. 2019; 13:629. https://doi.org/10.3389/fnins.2019.00629 [PubMed]
- 33. Mattson MP, Arumugam TV. Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab. 2018; 27:1176–99. https://doi.org/10.1016/j.cmet.2018.05.011 [PubMed]
- 34. Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, Craft S, Gandy S, Buettner C, Stoeckel LE, Holtzman DM, Nathan DM. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018; 14:168–81. https://doi.org/10.1038/nrneurol.2017.185 [PubMed]
- 35. Reid MA, Sanderson SM, Locasale JW. (2020). Cancer Metabolism. (Philadelphia). https://doi.org/10.1016/B978-0-323-47674-4.00009-8
- 36. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016; 2:e1600200. https://doi.org/10.1126/sciadv.1600200 [PubMed]
- 37. Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, Murphy CJ, Pauli C, Morris R, Taylor S, Bosch K, Yang S, Wang Y, Van Riper J, et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science. 2019; 363:1345–49. https://doi.org/10.1126/science.aat8515 [PubMed]
- 38. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008; 134:703–07. https://doi.org/10.1016/j.cell.2008.08.021 [PubMed]
- 39. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–33. https://doi.org/10.1126/science.1160809 [PubMed]
- 40. Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011; 14:443–51. https://doi.org/10.1016/j.cmet.2011.07.014 [PubMed]
- 41. Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012; 13:270–76. https://doi.org/10.1038/nrm3305 [PubMed]
- 42. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, Johnson J, Gatenby RA, Gillies RJ. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013; 73:1524–35. https://doi.org/10.1158/0008-5472.CAN-12-2796 [PubMed]
- 43. Forster JC, Harriss-Phillips WM, Douglass MJ, Bezak E. A review of the development of tumor vasculature and its effects on the tumor microenvironment. Hypoxia (Auckl). 2017; 5:21–32. https://doi.org/10.2147/HP.S133231 [PubMed]
- 44. Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat Rev. 2011; 37:63–74. https://doi.org/10.1016/j.ctrv.2010.05.001 [PubMed]
- 45. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004; 64:3892–99. https://doi.org/10.1158/0008-5472.CAN-03-2904 [PubMed]
- 46. Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, Lin HC, Yancopoulos GD, Glass DJ. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem. 2005; 280:2737–44. https://doi.org/10.1074/jbc.M407517200 [PubMed]
- 47. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017; 170:605–35. https://doi.org/10.1016/j.cell.2017.07.029 [PubMed]
- 48. Osborne CK, Bolan G, Monaco ME, Lippman ME. Hormone responsive human breast cancer in long-term tissue culture: effect of insulin. Proc Natl Acad Sci USA. 1976; 73:4536–40. https://doi.org/10.1073/pnas.73.12.4536 [PubMed]
- 49. Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL, Gaziano JM, Pollak M, Stampfer MJ. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol. 2008; 9:1039–47. https://doi.org/10.1016/S1470-2045(08)70235-3 [PubMed]
- 50. Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, Hartwick W, Hoffman B, Hood N. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol. 2002; 20:42–51. https://doi.org/10.1200/JCO.2002.20.1.42 [PubMed]
- 51. Orgel E, Mittelman SD. The links between insulin resistance, diabetes, and cancer. Curr Diab Rep. 2013; 13:213–22. https://doi.org/10.1007/s11892-012-0356-6 [PubMed]
- 52. Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y, Carboni JM, Gottardis MM, Pennisi PA, Molinolo AA, Kurshan N, Mejia W, Santopietro S, et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res. 2010; 70:741–51. https://doi.org/10.1158/0008-5472.CAN-09-2141 [PubMed]
- 53. Nencioni A, Caffa I, Cortellino S, Longo VD. Fasting and cancer: molecular mechanisms and clinical application. Nat Rev Cancer. 2018; 18:707–19. https://doi.org/10.1038/s41568-018-0061-0 [PubMed]
- 54. Tomas NM, Masur K, Piecha JC, Niggemann B, Zänker KS. Akt and phospholipase Cγ are involved in the regulation of growth and migration of MDA-MB-468 breast cancer and SW480 colon cancer cells when cultured with diabetogenic levels of glucose and insulin. BMC Res Notes. 2012; 5:214. https://doi.org/10.1186/1756-0500-5-214 [PubMed]
- 55. Nam S, Gupta VK, Lee HP, Lee JY, Wisdom KM, Varma S, Flaum EM, Davis C, West RB, Chaudhuri O. Cell cycle progression in confining microenvironments is regulated by a growth-responsive TRPV4-PI3K/Akt-p27Kip1 signaling axis. Sci Adv. 2019; 5:eaaw6171. https://doi.org/10.1126/sciadv.aaw6171 [PubMed]
- 56. Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003; 17:590–603. https://doi.org/10.1038/sj.leu.2402824 [PubMed]
- 57. Dossou AS, Basu A. The emerging roles of mTORC1 in macromanaging autophagy. Cancers (Basel). 2019; 11:1422. https://doi.org/10.3390/cancers11101422 [PubMed]
- 58. Paquette M, El-Houjeiri L, Pause A. mTOR pathways in cancer and autophagy. Cancers (Basel). 2018; 10:18. https://doi.org/10.3390/cancers10010018 [PubMed]
- 59. Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019; 9:1167–81. https://doi.org/10.1158/2159-8290.CD-19-0292 [PubMed]
- 60. Tian T, Li X, Zhang J. mTOR signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. Int J Mol Sci. 2019; 20:755. https://doi.org/10.3390/ijms20030755 [PubMed]
- 61. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014; 370:997–1007. https://doi.org/10.1056/NEJMoa1315226 [PubMed]
- 62. Miller BW, Przepiorka D, de Claro RA, Lee K, Nie L, Simpson N, Gudi R, Saber H, Shord S, Bullock J, Marathe D, Mehrotra N, Hsieh LS, et al. FDA approval: idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin Cancer Res. 2015; 21:1525–29. https://doi.org/10.1158/1078-0432.CCR-14-2522 [PubMed]
- 63. Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, Flinn IW, Flowers CR, Martin P, Viardot A, Blum KA, Goy AH, Davies AJ, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014; 370:1008–18. https://doi.org/10.1056/NEJMoa1314583 [PubMed]
- 64. Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, Friedman L, Turner M, Okkenhaug K, Vanhaesebroeck B. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014; 510:407–11. https://doi.org/10.1038/nature13444 [PubMed]
- 65. Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X, Wrasidlo W, Blair SL, Ginsberg MH, Cheresh DA, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011; 19:715–27. https://doi.org/10.1016/j.ccr.2011.04.016 [PubMed]
- 66. González-García A, Sánchez-Ruiz J, Flores JM, Carrera AC. Phosphatidylinositol 3-kinase gamma inhibition ameliorates inflammation and tumor growth in a model of colitis-associated cancer. Gastroenterology. 2010; 138:1374–83. https://doi.org/10.1053/j.gastro.2009.12.001 [PubMed]
- 67. Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015; 86:883–901. https://doi.org/10.1016/j.neuron.2015.03.035 [PubMed]
- 68. Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001; 21:1133–45. https://doi.org/10.1097/00004647-200110000-00001 [PubMed]
- 69. Yu Y, Herman P, Rothman DL, Agarwal D, Hyder F. Evaluating the gray and white matter energy budgets of human brain function. J Cereb Blood Flow Metab. 2018; 38:1339–53. https://doi.org/10.1177/0271678X17708691 [PubMed]
- 70. Mink JW, Blumenschine RJ, Adams DB. Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Physiol. 1981; 241:R203–12. https://doi.org/10.1152/ajpregu.1981.241.3.R203 [PubMed]
- 71. Magistretti PJ, Sorg O, Yu N, Martin JL, Pellerin L. Neurotransmitters regulate energy metabolism in astrocytes: implications for the metabolic trafficking between neural cells. Dev Neurosci. 1993; 15:306–12. https://doi.org/10.1159/000111349 [PubMed]
- 72. Hertz L, Dringen R, Schousboe A, Robinson SR. Astrocytes: glutamate producers for neurons. J Neurosci Res. 1999; 57:417–28. [PubMed]
- 73. Massucci FA, DiNuzzo M, Giove F, Maraviglia B, Castillo IP, Marinari E, De Martino A. Energy metabolism and glutamate-glutamine cycle in the brain: a stoichiometric modeling perspective. BMC Syst Biol. 2013; 7:103. https://doi.org/10.1186/1752-0509-7-103 [PubMed]
- 74. Mason S. Lactate shuttles in neuroenergetics-homeostasis, allostasis and beyond. Front Neurosci. 2017; 11:43. https://doi.org/10.3389/fnins.2017.00043 [PubMed]
- 75. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS, Woo PJ, Malenka RC, Vogel H, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell. 2015; 161:803–16. https://doi.org/10.1016/j.cell.2015.04.012 [PubMed]
- 76. Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, Körber C, Kardorff M, Ratliff M, Xie R, Horstmann H, Messer M, Paik SP, et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019; 573:532–38. https://doi.org/10.1038/s41586-019-1564-x [PubMed]
- 77. Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, McCabe BD, Galván JA, Robinson HP, Zlobec I, Ciriello G, Hanahan D. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 2019; 573:526–31. https://doi.org/10.1038/s41586-019-1576-6 [PubMed]
- 78. Vardjan N, Chowdhury HH, Horvat A, Velebit J, Malnar M, Muhič M, Kreft M, Krivec ŠG, Bobnar ST, Miš K, Pirkmajer S, Offermanns S, Henriksen G, et al. Enhancement of astroglial aerobic glycolysis by extracellular lactate-mediated increase in cAMP. Front Mol Neurosci. 2018; 11:148. https://doi.org/10.3389/fnmol.2018.00148 [PubMed]
- 79. Supplie LM, Düking T, Campbell G, Diaz F, Moraes CT, Götz M, Hamprecht B, Boretius S, Mahad D, Nave KA. Respiration-deficient astrocytes survive as glycolytic cells In vivo. J Neurosci. 2017; 37:4231–42. https://doi.org/10.1523/JNEUROSCI.0756-16.2017 [PubMed]
- 80. Yellen G. Fueling thought: management of glycolysis and oxidative phosphorylation in neuronal metabolism. J Cell Biol. 2018; 217:2235–46. https://doi.org/10.1083/jcb.201803152 [PubMed]
- 81. Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, Diaz F, Meijer D, Suter U, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012; 485:517–21. https://doi.org/10.1038/nature11007 [PubMed]
- 82. Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012; 487:443–48. https://doi.org/10.1038/nature11314 [PubMed]
- 83. Meyer N, Richter N, Fan Z, Siemonsmeier G, Pivneva T, Jordan P, Steinhäuser C, Semtner M, Nolte C, Kettenmann H. Oligodendrocytes in the mouse corpus callosum maintain axonal function by delivery of glucose. Cell Rep. 2018; 22:2383–94. https://doi.org/10.1016/j.celrep.2018.02.022 [PubMed]
- 84. Alcantara Llaguno SR, Wang Z, Sun D, Chen J, Xu J, Kim E, Hatanpaa KJ, Raisanen JM, Burns DK, Johnson JE, Parada LF. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell. 2015; 28:429–40. https://doi.org/10.1016/j.ccell.2015.09.007 [PubMed]
- 85. Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ, Um JY, Kim WK, Lee JK, Park J, Kim EH, Lee JH, Lee JH, et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature. 2018; 560:243–47. https://doi.org/10.1038/s41586-018-0389-3 [PubMed]
- 86. Willis CM, Nicaise AM, Bongarzone ER, Givogri M, Reiter CR, Heintz O, Jellison ER, Sutter PA, TeHennepe G, Ananda G, Vella AT, Crocker SJ. Astrocyte support for oligodendrocyte differentiation can be conveyed via extracellular vesicles but diminishes with age. Sci Rep. 2020; 10:828. https://doi.org/10.1038/s41598-020-57663-x [PubMed]
- 87. Brix B, Mesters JR, Pellerin L, Jöhren O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1α-mediated target gene activation. J Neurosci. 2012; 32:9727–35. https://doi.org/10.1523/JNEUROSCI.0879-12.2012 [PubMed]
- 88. Heni M, Hennige AM, Peter A, Siegel-Axel D, Ordelheide AM, Krebs N, Machicao F, Fritsche A, Häring HU, Staiger H. Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS One. 2011; 6:e21594. https://doi.org/10.1371/journal.pone.0021594 [PubMed]
- 89. Goebbels S, Oltrogge JH, Kemper R, Heilmann I, Bormuth I, Wolfer S, Wichert SP, Möbius W, Liu X, Lappe-Siefke C, Rossner MJ, Groszer M, Suter U, et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J Neurosci. 2010; 30:8953–64. https://doi.org/10.1523/JNEUROSCI.0219-10.2010 [PubMed]
- 90. Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008; 28:7174–83. https://doi.org/10.1523/JNEUROSCI.0150-08.2008 [PubMed]
- 91. Arai K, Lo EH. Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J Neurosci Res. 2010; 88:758–63. https://doi.org/10.1002/jnr.22256 [PubMed]
- 92. Strickland M, Stoll EA. Metabolic reprogramming in glioma. Front Cell Dev Biol. 2017; 5:43. https://doi.org/10.3389/fcell.2017.00043 [PubMed]
- 93. Zhou W, Wahl DR. Metabolic abnormalities in glioblastoma and metabolic strategies to overcome treatment resistance. Cancers (Basel). 2019; 11:1231. https://doi.org/10.3390/cancers11091231 [PubMed]
- 94. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–68. https://doi.org/10.1038/nature07385 [PubMed]
- 95. Suzuki Y, Shirai K, Oka K, Mobaraki A, Yoshida Y, Noda SE, Okamoto M, Suzuki Y, Itoh J, Itoh H, Ishiuchi S, Nakano T. Higher pAkt expression predicts a significant worse prognosis in glioblastomas. J Radiat Res. 2010; 51:343–48. https://doi.org/10.1269/jrr.09109 [PubMed]
- 96. Bai H, Harmanci AS, Erson-Omay EZ, Li J, Coşkun S, Simon M, Krischek B, Özduman K, Omay SB, Sorensen EA, Turcan Ş, Bakirciğlu M, Carrión-Grant G, et al. Integrated genomic characterization of IDH1-mutant glioma Malignant progression. Nat Genet. 2016; 48:59–66. https://doi.org/10.1038/ng.3457 [PubMed]
- 97. Wen PY, Touat M, Alexander BM, Mellinghoff IK, Ramkissoon S, McCluskey CS, Pelton K, Haidar S, Basu SS, Gaffey SC, Brown LE, Martinez-Ledesma JE, Wu S, et al. Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3-kinase pathway activation: an open-label, multicenter, multi-arm, phase II trial. J Clin Oncol. 2019; 37:741–50. https://doi.org/10.1200/JCO.18.01207 [PubMed]
- 98. Liu P, Zhu C, Luo J, Lan S, Su D, Wang Q, Wei Z, Cui W, Xu C, Yang X. Par6 regulates cell cycle progression through enhancement of Akt/PI3K/GSK-3β signaling pathway activation in glioma. FASEB J. 2020; 34:1481–96. https://doi.org/10.1096/fj.201901629RR [PubMed]
- 99. Tateishi K, Nakamura T, Juratli TA, Williams EA, Matsushita Y, Miyake S, Nishi M, Miller JJ, Tummala SS, Fink AL, Lelic N, Koerner MV, Miyake Y, et al. PI3K/AKT/mTOR pathway alterations promote Malignant progression and xenograft formation in oligodendroglial tumors. Clin Cancer Res. 2019; 25:4375–87. https://doi.org/10.1158/1078-0432.CCR-18-4144 [PubMed]
- 100. Gong Y, Ma Y, Sinyuk M, Loganathan S, Thompson RC, Sarkaria JN, Chen W, Lathia JD, Mobley BC, Clark SW, Wang J. Insulin-mediated signaling promotes proliferation and survival of glioblastoma through Akt activation. Neuro Oncol. 2016; 18:48–57. https://doi.org/10.1093/neuonc/nov096 [PubMed]
- 101. Li Y, He ZC, Liu Q, Zhou K, Shi Y, Yao XH, Zhang X, Kung HF, Ping YF, Bian XW. Large intergenic non-coding RNA-RoR inhibits aerobic glycolysis of glioblastoma cells via Akt pathway. J Cancer. 2018; 9:880–89. https://doi.org/10.7150/jca.20869 [PubMed]
- 102. Langhans J, Schneele L, Trenkler N, von Bandemer H, Nonnenmacher L, Karpel-Massler G, Siegelin MD, Zhou S, Halatsch ME, Debatin KM, Westhoff MA. The effects of PI3K-mediated signalling on glioblastoma cell behaviour. Oncogenesis. 2017; 6:398. https://doi.org/10.1038/s41389-017-0004-8 [PubMed]
- 103. Daniel PM, Filiz G, Brown DV, Christie M, Waring PM, Zhang Y, Haynes JM, Pouton C, Flanagan D, Vincan E, Johns TG, Montgomery K, Phillips WA, Mantamadiotis T. PI3K activation in neural stem cells drives tumorigenesis which can be ameliorated by targeting the cAMP response element binding protein. Neuro Oncol. 2018; 20:1344–55. https://doi.org/10.1093/neuonc/noy068 [PubMed]
- 104. Wang YJ, Chang SB, Wang CY, Huang HT, Tzeng SF. The selective lipoprotein-associated phospholipase A2 inhibitor darapladib triggers irreversible actions on glioma cell apoptosis and mitochondrial dysfunction. Toxicol Appl Pharmacol. 2020; 402:115133. https://doi.org/10.1016/j.taap.2020.115133 [PubMed]
- 105. Chen RQ, Xu XH, Liu F, Li CY, Li YJ, Li XR, Jiang GY, Hu F, Liu D, Pan F, Qiu XY, Chen XQ. The binding of PD-L1 and Akt facilitates glioma cell invasion upon starvation via Akt/Autophagy/F-Actin signaling. Front Oncol. 2019; 9:1347. https://doi.org/10.3389/fonc.2019.01347 [PubMed]
- 106. Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA, Debnath J, Shokat KM, Weiss WA. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010; 3:ra81. https://doi.org/10.1126/scisignal.2001017 [PubMed]
- 107. Ellert-Miklaszewska A, Dabrowski M, Lipko M, Sliwa M, Maleszewska M, Kaminska B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia. 2013; 61:1178–90. https://doi.org/10.1002/glia.22510 [PubMed]
- 108. Lisi L, Laudati E, Navarra P, Dello Russo C. The mTOR kinase inhibitors polarize glioma-activated microglia to express a M1 phenotype. J Neuroinflammation. 2014; 11:125. https://doi.org/10.1186/1742-2094-11-125 [PubMed]
- 109. Wei J, Gabrusiewicz K, Heimberger A. The controversial role of microglia in Malignant gliomas. Clin Dev Immunol. 2013; 2013:285246. https://doi.org/10.1155/2013/285246 [PubMed]
- 110. Oh T, Ivan ME, Sun MZ, Safaee M, Fakurnejad S, Clark AJ, Sayegh ET, Bloch O, Parsa AT. PI3K pathway inhibitors: potential prospects as adjuncts to vaccine immunotherapy for glioblastoma. Immunotherapy. 2014; 6:737–53. https://doi.org/10.2217/imt.14.35 [PubMed]
- 111. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, Segall JE. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012; 18:519–27. https://doi.org/10.2119/molmed.2011.00217 [PubMed]
- 112. Sartorius T, Peter A, Heni M, Maetzler W, Fritsche A, Häring HU, Hennige AM. The brain response to peripheral insulin declines with age: a contribution of the blood-brain barrier? PLoS One. 2015; 10:e0126804. https://doi.org/10.1371/journal.pone.0126804 [PubMed]
- 113. Stanley M, Macauley SL, Holtzman DM. Changes in insulin and insulin signaling in Alzheimer’s disease: cause or consequence? J Exp Med. 2016; 213:1375–85. https://doi.org/10.1084/jem.20160493 [PubMed]
- 114. Frölich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Türk A, Hoyer S, Zöchling R, Boissl KW, Jellinger K, Riederer P. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm (Vienna). 1998; 105:423–38. https://doi.org/10.1007/s007020050068 [PubMed]
- 115. Tong M, Dong M, de la Monte SM. Brain insulin-like growth factor and neurotrophin resistance in Parkinson’s disease and dementia with lewy bodies: potential role of manganese neurotoxicity. J Alzheimers Dis. 2009; 16:585–99. https://doi.org/10.3233/JAD-2009-0995 [PubMed]
- 116. Oka M, Suzuki E, Asada A, Saito T, Iijima KM, Ando K. Increasing glucose uptake into neurons antagonizes brain aging and promotes health and life span under dietary restriction in Drosophila. bioRxiv. 2020. https://doi.org/10.1101/2020.06.11.145227
- 117. Ding F, Yao J, Rettberg JR, Chen S, Brinton RD. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One. 2013; 8:e79977. https://doi.org/10.1371/journal.pone.0079977 [PubMed]
- 118. Castelli V, Benedetti E, Antonosante A, Catanesi M, Pitari G, Ippoliti R, Cimini A, d’Angelo M. Neuronal cells rearrangement during aging and neurodegenerative disease: metabolism, oxidative stress and organelles dynamic. Front Mol Neurosci. 2019; 12:132. https://doi.org/10.3389/fnmol.2019.00132 [PubMed]
- 119. Chow HM, Shi M, Cheng A, Gao Y, Chen G, Song X, So RW, Zhang J, Herrup K. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat Neurosci. 2019; 22:1806–19. https://doi.org/10.1038/s41593-019-0505-1 [PubMed]
- 120. Goyal MS, Vlassenko AG, Blazey TM, Su Y, Couture LE, Durbin TJ, Bateman RJ, Benzinger TL, Morris JC, Raichle ME. Loss of brain Aerobic glycolysis in normal human aging. Cell Metab. 2017; 26:353–60.e3. https://doi.org/10.1016/j.cmet.2017.07.010 [PubMed]
- 121. de la Torre JC. Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesis. Neurobiol Aging. 2000; 21:331–42. https://doi.org/10.1016/s0197-4580(00)00111-1 [PubMed]
- 122. Almeida A, Moncada S, Bolaños JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004; 6:45–51. https://doi.org/10.1038/ncb1080 [PubMed]
- 123. Fernandez AM, Hernandez-Garzón E, Perez-Domper P, Perez-Alvarez A, Mederos S, Matsui T, Santi A, Trueba-Saiz A, García-Guerra L, Pose-Utrilla J, Fielitz J, Olson EN, Fernandez de la Rosa R, et al. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes. 2017; 66:64–74. https://doi.org/10.2337/db16-0861 [PubMed]
- 124. Zuendorf G, Kerrouche N, Herholz K, Baron JC. Efficient principal component analysis for multivariate 3D voxel-based mapping of brain functional imaging data sets as applied to FDG-PET and normal aging. Hum Brain Mapp. 2003; 18:13–21. https://doi.org/10.1002/hbm.10069 [PubMed]
- 125. Demetrius LA, Magistretti PJ, Pellerin L. Alzheimer’s disease: the amyloid hypothesis and the inverse Warburg effect. Front Physiol. 2015; 5:522. https://doi.org/10.3389/fphys.2014.00522 [PubMed]
- 126. Girault FM, Sonnay S, Gruetter R, Duarte JM. Alterations of brain energy metabolism in type 2 diabetic Goto-Kakizaki rats measured in vivo by13 C magnetic resonance spectroscopy. Neurotox Res. 2019; 36:268–78. https://doi.org/10.1007/s12640-017-9821-y [PubMed]
- 127. García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, Jastroch M, Johansson P, Ninkovic J, Yi CX, Le Thuc O, Szigeti-Buck K, Cai W, et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell. 2016; 166:867–80. https://doi.org/10.1016/j.cell.2016.07.028 [PubMed]
- 128. Li W, Roy Choudhury G, Winters A, Prah J, Lin W, Liu R, Yang SH. Hyperglycemia alters astrocyte metabolism and inhibits astrocyte proliferation. Aging Dis. 2018; 9:674–84. https://doi.org/10.14336/AD.2017.1208 [PubMed]
- 129. Gorgisen G, Yaren Z. Insulin receptor substrate 1 overexpression promotes survival of glioblastoma cells through AKT1 activation. Folia Neuropathol. 2020; 58:38–44. https://doi.org/10.5114/fn.2020.94005 [PubMed]
- 130. Almiron Bonnin DA, Ran C, Havrda MC, Liu H, Hitoshi Y, Zhang Z, Cheng C, Ung M, Israel MA. Insulin-mediated signaling facilitates resistance to PDGFR inhibition in proneural hPDGFB-driven gliomas. Mol Cancer Ther. 2017; 16:705–16. https://doi.org/10.1158/1535-7163.MCT-16-0616 [PubMed]
- 131. Cuevas-Diaz Duran R, Wang CY, Zheng H, Deneen B, Wu JQ. Brain region-specific gene signatures revealed by distinct astrocyte subpopulations unveil links to glioma and neurodegenerative diseases. eNeuro. 2019; 6. https://doi.org/10.1523/ENEURO.0288-18.2019 [PubMed]
- 132. Irvin DM, McNeill RS, Bash RE, Miller CR. Intrinsic astrocyte heterogeneity influences tumor growth in glioma mouse models. Brain Pathol. 2017; 27:36–50. https://doi.org/10.1111/bpa.12348 [PubMed]
- 133. Xing F, Luan Y, Cai J, Wu S, Mai J, Gu J, Zhang H, Li K, Lin Y, Xiao X, Liang J, Li Y, Chen W, et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1α Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017; 18:468–81. https://doi.org/10.1016/j.celrep.2016.12.037 [PubMed]
- 134. Zhang H, Zhou Y, Cui B, Liu Z, Shen H. Novel insights into astrocyte-mediated signaling of proliferation, invasion and tumor immune microenvironment in glioblastoma. Biomed Pharmacother. 2020; 126:110086. https://doi.org/10.1016/j.biopha.2020.110086 [PubMed]
- 135. Cheng CK, Fan QW, Weiss WA. PI3K signaling in glioma—animal models and therapeutic challenges. Brain Pathol. 2009; 19:112–20. https://doi.org/10.1111/j.1750-3639.2008.00233.x [PubMed]
- 136. Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009; 4:14. https://doi.org/10.1186/1750-1326-4-14 [PubMed]
- 137. Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014; 26:2694–701. https://doi.org/10.1016/j.cellsig.2014.08.019 [PubMed]
- 138. Martínez-Mármol R, Mohannak N, Qian L, Wang T, Gormal RS, Ruitenberg MJ, Vanhaesebroeck B, Coulson EJ, Meunier FA. P110δ PI3-kinase inhibition perturbs APP and TNFα trafficking, reduces plaque burden, dampens neuroinflammation, and prevents cognitive decline in an Alzheimer’s disease mouse model. J Neurosci. 2019; 39:7976–91. https://doi.org/10.1523/JNEUROSCI.0674-19.2019 [PubMed]
- 139. Wongrakpanich S, Hurst A, Bustamante J, Candelario N, Biso S, Chaiwatcharayut W, Dourado C, Rosenzweig A. Prognostic significance of dementia in older adults with solid tumors. Dement Geriatr Cogn Disord. 2017; 43:38–44. https://doi.org/10.1159/000453449 [PubMed]
- 140. Baillargeon J, Kuo YF, Lin YL, Raji MA, Singh A, Goodwin JS. Effect of mental disorders on diagnosis, treatment, and survival of older adults with colon cancer. J Am Geriatr Soc. 2011; 59:1268–73. https://doi.org/10.1111/j.1532-5415.2011.03481.x [PubMed]
- 141. Gorin SS, Heck JE, Albert S, Hershman D. Treatment for breast cancer in patients with Alzheimer’s disease. J Am Geriatr Soc. 2005; 53:1897–904. https://doi.org/10.1111/j.1532-5415.2005.00467.x [PubMed]
- 142. Raji MA, Kuo YF, Freeman JL, Goodwin JS. Effect of a dementia diagnosis on survival of older patients after a diagnosis of breast, colon, or prostate cancer: implications for cancer care. Arch Intern Med. 2008; 168:2033–40. https://doi.org/10.1001/archinte.168.18.2033 [PubMed]
- 143. Gurney J, Sarfati D, Stanley J. The impact of patient comorbidity on cancer stage at diagnosis. Br J Cancer. 2015; 113:1375–80. https://doi.org/10.1038/bjc.2015.355 [PubMed]
- 144. Musicco M, Adorni F, Di Santo S, Prinelli F, Pettenati C, Caltagirone C, Palmer K, Russo A. Inverse occurrence of cancer and Alzheimer disease: a population-based incidence study. Neurology. 2013; 81:322–28. https://doi.org/10.1212/WNL.0b013e31829c5ec1 [PubMed]
- 145. Ou SM, Lee YJ, Hu YW, Liu CJ, Chen TJ, Fuh JL, Wang SJ. Does Alzheimer’s disease protect against cancers? A nationwide population-based study. Neuroepidemiology. 2013; 40:42–49. https://doi.org/10.1159/000341411 [PubMed]
- 146. McWilliams L. An overview of treating people with comorbid dementia: implications for cancer care. Clin Oncol (R Coll Radiol). 2020; 32:562–68. https://doi.org/10.1016/j.clon.2020.06.014 [PubMed]
- 147. van der Willik KD, Schagen SB, Ikram MA. Cancer and dementia: two sides of the same coin? Eur J Clin Invest. 2018; 48:e13019. https://doi.org/10.1111/eci.13019 [PubMed]
- 148. Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 2018; 22:269–85. https://doi.org/10.1016/j.celrep.2017.12.039 [PubMed]
- 149. Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA. 2018; 115:E1896–905. https://doi.org/10.1073/pnas.1800165115 [PubMed]
- 150. Pan J, Ma N, Yu B, Zhang W, Wan J. Transcriptomic profiling of microglia and astrocytes throughout aging. J Neuroinflammation. 2020; 17:97. https://doi.org/10.1186/s12974-020-01774-9 [PubMed]
- 151. Galvão RP, Zong H. Inflammation and gliomagenesis: Bi-directional communication at early and late stages of tumor progression. Curr Pathobiol Rep. 2013; 1:19–28. https://doi.org/10.1007/s40139-012-0006-3 [PubMed]
- 152. De Boeck A, Ahn BY, D’Mello C, Lun X, Menon SV, Alshehri MM, Szulzewsky F, Shen Y, Khan L, Dang NH, Reichardt E, Goring KA, King J, et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat Commun. 2020; 11:4997. https://doi.org/10.1038/s41467-020-18569-4 [PubMed]
- 153. Salazar-Ramiro A, Ramírez-Ortega D, Pérez de la Cruz V, Hérnandez-Pedro NY, González-Esquivel DF, Sotelo J, Pineda B. Role of redox status in development of glioblastoma. Front Immunol. 2016; 7:156. https://doi.org/10.3389/fimmu.2016.00156 [PubMed]
- 154. Bellaver B, Souza DG, Souza DO, Quincozes-Santos A. Hippocampal astrocyte cultures from adult and aged rats reproduce changes in glial functionality observed in the aging brain. Mol Neurobiol. 2017; 54:2969–85. https://doi.org/10.1007/s12035-016-9880-8 [PubMed]
- 155. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004; 432:396–401. https://doi.org/10.1038/nature03128 [PubMed]
- 156. Koso H, Takeda H, Yew CC, Ward JM, Nariai N, Ueno K, Nagasaki M, Watanabe S, Rust AG, Adams DJ, Copeland NG, Jenkins NA. Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proc Natl Acad Sci USA. 2012; 109:E2998–3007. https://doi.org/10.1073/pnas.1215899109 [PubMed]
- 157. Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, Cortes-Ciriano I, Birzu C, Geduldig JE, Pelton K, Lim-Fat MJ, Pal S, Ferrer-Luna R, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. 2020; 580:517–23. https://doi.org/10.1038/s41586-020-2209-9 [PubMed]
- 158. McCord M, Steffens A, Javier R, Kam KL, McCortney K, Horbinski C. The efficacy of DNA mismatch repair enzyme immunohistochemistry as a screening test for hypermutated gliomas. Acta Neuropathol Commun. 2020; 8:15. https://doi.org/10.1186/s40478-020-0892-2 [PubMed]
- 159. Daniel P, Sabri S, Chaddad A, Meehan B, Jean-Claude B, Rak J, Abdulkarim BS. Temozolomide induced hypermutation in glioma: evolutionary mechanisms and therapeutic opportunities. Front Oncol. 2019; 9:41. https://doi.org/10.3389/fonc.2019.00041 [PubMed]
- 160. Abedalthagafi M, Barakeh D, Foshay KM. Immunogenetics of glioblastoma: the future of personalized patient management. NPJ Precis Oncol. 2018; 2:27. https://doi.org/10.1038/s41698-018-0070-1 [PubMed]
- 161. Shen Y, Grisdale CJ, Islam SA, Bose P, Lever J, Zhao EY, Grinshtein N, Ma Y, Mungall AJ, Moore RA, Lun X, Senger DL, Robbins SM, et al. Comprehensive genomic profiling of glioblastoma tumors, BTICs, and xenografts reveals stability and adaptation to growth environments. Proc Natl Acad Sci USA. 2019; 116:19098–108. https://doi.org/10.1073/pnas.1813495116 [PubMed]
- 162. Noch E, Khalili K. Oncogenic viruses and tumor glucose metabolism: like kids in a candy store. Mol Cancer Ther. 2012; 11:14–23. https://doi.org/10.1158/1535-7163.MCT-11-0517 [PubMed]
- 163. Helt AM, Galloway DA. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis. 2003; 24:159–69. https://doi.org/10.1093/carcin/24.2.159 [PubMed]
- 164. Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. 2015; 350:568–71. https://doi.org/10.1126/science.aab3291 [PubMed]
- 165. Gilden DH, Lipton HL. (2012). Clinical and molecular aspects of neurotropic virus infection: Elsevier Inc.).
- 166. Lee K, Jeon K, Kim JM, Kim VN, Choi DH, Kim SU, Kim S. Downregulation of GFAP, TSP-1, and p53 in human glioblastoma cell line, U373MG, by IE1 protein from human cytomegalovirus. Glia. 2005; 51:1–12. https://doi.org/10.1002/glia.20179 [PubMed]
- 167. Lilyestrom W, Klein MG, Zhang R, Joachimiak A, Chen XS. Crystal structure of SV40 large T-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev. 2006; 20:2373–82. https://doi.org/10.1101/gad.1456306 [PubMed]
- 168. Greenway AL, McPhee DA, Allen K, Johnstone R, Holloway G, Mills J, Azad A, Sankovich S, Lambert P. Human immunodeficiency virus type 1 Nef binds to tumor suppressor p53 and protects cells against p53-mediated apoptosis. J Virol. 2002; 76:2692–702. https://doi.org/10.1128/jvi.76.6.2692-2702.2002 [PubMed]
- 169. Caracciolo V, Macaluso M, D’Agostino L, Montanari M, Scheff J, Reiss K, Khalili K, Giordano A. Cross-talk between T-Ag presence and pRb family and p53/p73 signaling in mouse and human medulloblastoma. J Cell Biochem. 2010; 110:182–90. https://doi.org/10.1002/jcb.22525 [PubMed]
- 170. Del Valle L, Gordon J, Assimakopoulou M, Enam S, Geddes JF, Varakis JN, Katsetos CD, Croul S, Khalili K. Detection of JC virus DNA sequences and expression of the viral regulatory protein T-antigen in tumors of the central nervous system. Cancer Res. 2001; 61:4287–93. [PubMed]
- 171. Noch E, Sariyer IK, Gordon J, Khalili K. JC virus T-antigen regulates glucose metabolic pathways in brain tumor cells. PLoS One. 2012; 7:e35054. https://doi.org/10.1371/journal.pone.0035054 [PubMed]
- 172. Krynska B, Gordon J, Otte J, Franks R, Knobler R, DeLuca A, Giordano A, Khalili K. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J Cell Biochem. 1997; 67:223–30. https://doi.org/10.1002/(sici)1097-4644(19971101)67:2<223::aid-jcb7>3.0.co;2-z [PubMed]
- 173. Khalili K, Del Valle L, Wang JY, Darbinian N, Lassak A, Safak M, Reiss K. T-antigen of human polyomavirus JC cooperates withIGF-IR signaling system in cerebellar tumors of the childhood-medulloblastomas. Anticancer Res. 2003; 23:2035–41. [PubMed]
- 174. Diehl N, Schaal H. Make yourself at home: viral hijacking of the PI3K/Akt signaling pathway. Viruses. 2013; 5:3192–212. https://doi.org/10.3390/v5123192 [PubMed]
- 175. Yu L, Chen X, Wang L, Chen S. Oncogenic virus-induced aerobic glycolysis and tumorigenesis. J Cancer. 2018; 9:3699–706. https://doi.org/10.7150/jca.27279 [PubMed]
- 176. Mao H, Lebrun DG, Yang J, Zhu VF, Li M. Deregulated signaling pathways in glioblastoma multiforme: molecular mechanisms and therapeutic targets. Cancer Invest. 2012; 30:48–56. https://doi.org/10.3109/07357907.2011.630050 [PubMed]
- 177. Regad T. Targeting RTK signaling pathways in cancer. Cancers (Basel). 2015; 7:1758–84. https://doi.org/10.3390/cancers7030860 [PubMed]
- 178. Mecca C, Giambanco I, Donato R, Arcuri C. Targeting mTOR in glioblastoma: rationale and preclinical/clinical evidence. Dis Markers. 2018; 2018:9230479. https://doi.org/10.1155/2018/9230479 [PubMed]
- 179. Koul D, Shen R, Bergh S, Sheng X, Shishodia S, Lafortune TA, Lu Y, de Groot JF, Mills GB, Yung WK. Inhibition of Akt survival pathway by a small-molecule inhibitor in human glioblastoma. Mol Cancer Ther. 2006; 5:637–44. https://doi.org/10.1158/1535-7163.MCT-05-0453 [PubMed]
- 180. Djuzenova CS, Fiedler V, Memmel S, Katzer A, Sisario D, Brosch PK, Göhrung A, Frister S, Zimmermann H, Flentje M, Sukhorukov VL. Differential effects of the Akt inhibitor MK-2206 on migration and radiation sensitivity of glioblastoma cells. BMC Cancer. 2019; 19:299. https://doi.org/10.1186/s12885-019-5517-4 [PubMed]
- 181. Verret B, Cortes J, Bachelot T, Andre F, Arnedos M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann Oncol. 2019; 30:x12–x20. https://doi.org/10.1093/annonc/mdz381
- 182. Batsios G, Viswanath P, Subramani E, Najac C, Gillespie AM, Santos RD, Molloy AR, Pieper RO, Ronen SM. PI3K/mTOR inhibition of IDH1 mutant glioma leads to reduced 2HG production that is associated with increased survival. Sci Rep. 2019; 9:10521. https://doi.org/10.1038/s41598-019-47021-x [PubMed]
- 183. Noch E, Yim I, Cantley L. PI3K inhibition in conjuction with the ketogenic diet reduces growth and neuroinflammation in the pediatric high grade glioma. Neuro Oncol. 2019; 21:vi89–vi90. https://doi.org/10.1093/neuonc/noz175.368
- 184. Shams-White MM, Brockton NT, Mitrou P, Romaguera D, Brown S, Bender A, Kahle LL, Reedy J. Operationalizing the 2018 world cancer research fund/American institute for cancer research (WCRF/AICR) cancer prevention recommendations: a standardized scoring system. Nutrients. 2019; 11:1572. https://doi.org/10.3390/nu11071572 [PubMed]
- 185. Brasure M, Desai P, Davila H, Nelson VA, Calvert C, Jutkowitz E, Butler M, Fink HA, Ratner E, Hemmy LS, McCarten JR, Barclay TR, Kane RL. Physical activity interventions in preventing cognitive decline and Alzheimer-type dementia: a systematic review. Ann Intern Med. 2018; 168:30–38. https://doi.org/10.7326/M17-1528 [PubMed]
- 186. Schelke MW, Hackett K, Chen JL, Shih C, Shum J, Montgomery ME, Chiang GC, Berkowitz C, Seifan A, Krikorian R, Isaacson RS. Nutritional interventions for Alzheimer’s prevention: a clinical precision medicine approach. Ann N Y Acad Sci. 2016; 1367:50–56. https://doi.org/10.1111/nyas.13070 [PubMed]