



Introduction
A severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of coronavirus disease 2019 (COVID-19). The virus emerged in Wuhan, China at the end of December 2019 and quickly spread to the world [1, 2]. On March 11, 2020, the World Health Organization (WHO) declared the COVID-19 outbreak a pandemic. The virus is highly infectious. As of October 23, 2020, COVID-19 has infected more than 60 million people, caused more than 1.4 million deaths, and the number of patients and deaths continues to increase, posing a serious health threat to the world. The symptoms of COVID-19 infection include high fever, cough and shortness of breath, and severe cases can lead to kidney failure and death [3, 4]. Currently, there are no approved drugs or vaccines against COVID-19.
SARS-CoV-2 belongs to the beta coronavirus group [5]. Generally, β-coronaviruses produce an 800-kDa polypeptides during genome transcription. The polypeptide is proteolytically cleaved to produce various proteins [2, 6]. The proteolytic process is mediated by papain-like protease (PLpro) and 3-chymotrypsin-like protease (3CLpro). 3CLpro cleaves polyprotein at 11 distinct sites to generate various non-structural proteins that are important for viral replication [7, 8]. In view of the key role of 3CLpro in viral replication, 3CLpro is used as a potential target against COVID-19 inhibitors.
Since the outbreak of COVID-19, researchers had obtained multiple structures of SARS-CoV-2 protein using X-ray diffraction or cryo-electron microscopy. The structure used in this study was the complex crystal structures of SARS-CoV-2 3CLpro protein combined with a synthetic peptidomimetic inhibitor N3 (PDB: 6LU7, Figure 1) [9]. The 3CLpro protein has three important domains I-III, corresponding to residues Phe8-Tyr101, Lys102-Pro184, and Thr201-Val303. There is also a connecting loop corresponding to residues Phe185-Ile200, which connects domains II and III. The 3CLpro protein has an important catalytic dyads consisting of His41 and Cys145, and the substrate-binding site is located in the cleft between domains I and II [9–11].
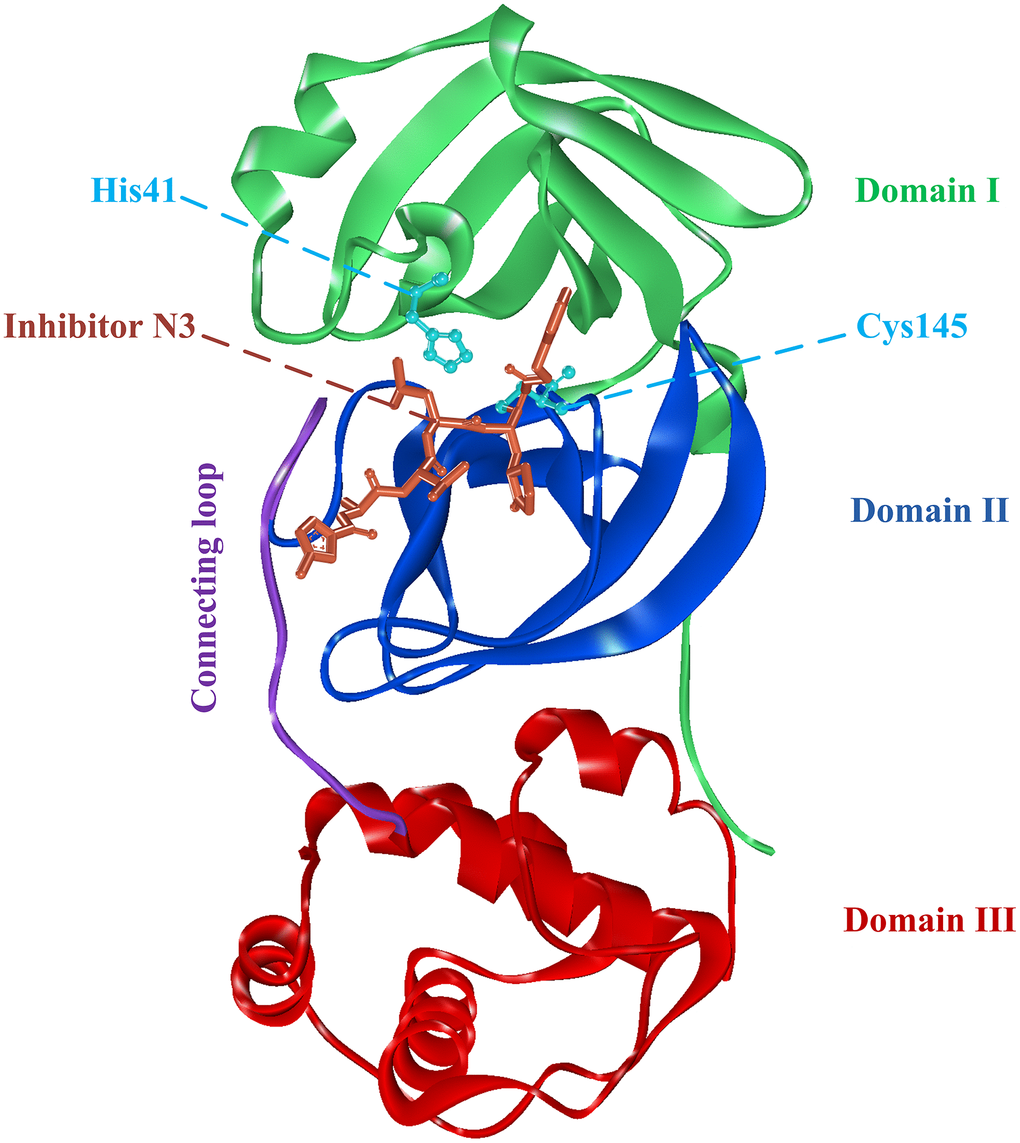
Figure 1. The complex structure of 3CLpro protein and synthetic peptidomimetic inhibitor N3. 3CLpro contains three domains, namely domain I, domain II, and domain III, and a connecting loop. Residues His41 and Cys145 are important catalytic dyads.
In this study, we used the SARS-COV-2 3CLpro protein as the target to conduct high-throughput virtual screening of the FDA-approved drugs, and sorted them according to the level of docking scores to find potential inhibitors for effective treatment of SARS-COV-2. Taking the inhibitor Indinavir with the highest docking score as a representative, we further explored the binding pattern of 3CLpro and Indinavir, and calculated the binding free energy between them to evaluate the binding affinity. Subsequently, in order to further investigate the effect of the Indinavir on the conformation of 3CLpro protein, 100 ns molecular dynamics (MD) simulations were performed for the 3CLpro system and the 3CLpro-Indinavir complex system, respectively, and post-dynamic analyses were performed. The stability of the two systems was evaluated by using root mean square deviation (RMSD) and root mean square fluctuation (RMSF). Principal component analysis (PCA) and dynamic cross-correlation diagram (DCCM) were used to explore the differences in the conformational state and correlated motions of the two systems. Through the analyses of the residue interaction network (RIN) and residue-residue hydrogen bond (H-bond) occupancy, the differences in the interaction between the two systems were explored, and the underlying reason for the conformational differences between the two systems were further revealed. Based on the above research, we hoped that this research would provide valuable information for the clinical treatment against COVID-19.
Results
Result on high-throughput virtual screening
Virtual screening was an effective method for the development of new drugs with desired characteristics and structural diversity [12]. In this study, through the virtual screening method based on docking, high-throughput virtual screening was performed on the FDA-approved drugs from the ZINC database. Before docking screening, in order to evaluate the reliability of the CDOCKER model, the ligand N3 in the original crystal structure was reinserted into the binding pocket. It was found that the RMSD between the docking conformation of the original ligand and the eutectic conformation was 0.8674Å (less than 2Å), indicating that the docking method and parameters were reliable. After that, all compounds were docked to the active site of 3CLpro protein and sorted according to the level of docking score. Through virtual screening, we had obtained a number of valuable drugs against COVID-19. Table 1 listed the top 10 drugs with the docking score. Among them, Indinavir, as an anti-HIV protease inhibitor, had the highest docking score with 3CLpro protein.
Table 1. Top 10 FDA-approved drugs with docking score based on high-throughput virtual screening.
| ID | Name | Chemical structure | Docking score |
| ZINC22448696 | Indinavir | 49.5328 | |
| ZINC3944422 | Ritonavir | 49.4166 | |
| ZINC3914596 | Saquinavir | 49.3129 | |
| ZINC3951740 | Lopinavir | 49.0524 | |
| ZINC85537014 | Cobicistat | 48.9127 | |
| ZINC100016058 | Tipranavir | 48.8735 | |
| ZINC3831151 | Montelukast | 48.8146 | |
| ZINC49841054 | Kyprolis | 48.8093 | |
| ZINC29571072 | Isavuconazonium | 47.9368 | |
| ZINC14879972 | Tigecycline | 47.9302 |
Investigation on the binding pattern
We chose the Indinavir to explore its binding pattern with 3CLpro protein. Figure 2A showed that Indinavir was docked to the binding site of the 3CLpro protein, Figure 2B showed the binding pocket of the Indinavir and protein, and Figure 2C showed a 2D diagram of the interaction between the Indinavir and protein. It could be seen from the 2D diagram that the Indinavir could form 7 stable H-bond interactions with residues His41, Gly143, Cys145, Glu166, Arg188, Thr190 and Gln192, and formed van der waals (VDW) interactions with residues Thr25, Leu27, Cys44, Ala46, Met49, Tyr54, Phe140, Leu141, His163, His164, Met165, Leu167, Pro168 and Asp187. It was worth noting that the conserved catalytic dyad residues His41 and Cys145 in the protein were tightly bound with the Indinavir, which was beneficial to stabilize the protein. Based on the multiple stable interactions formed between the protein and the ligand, the Indinavir could play a role in inhibiting the activity of 3CLpro protein.
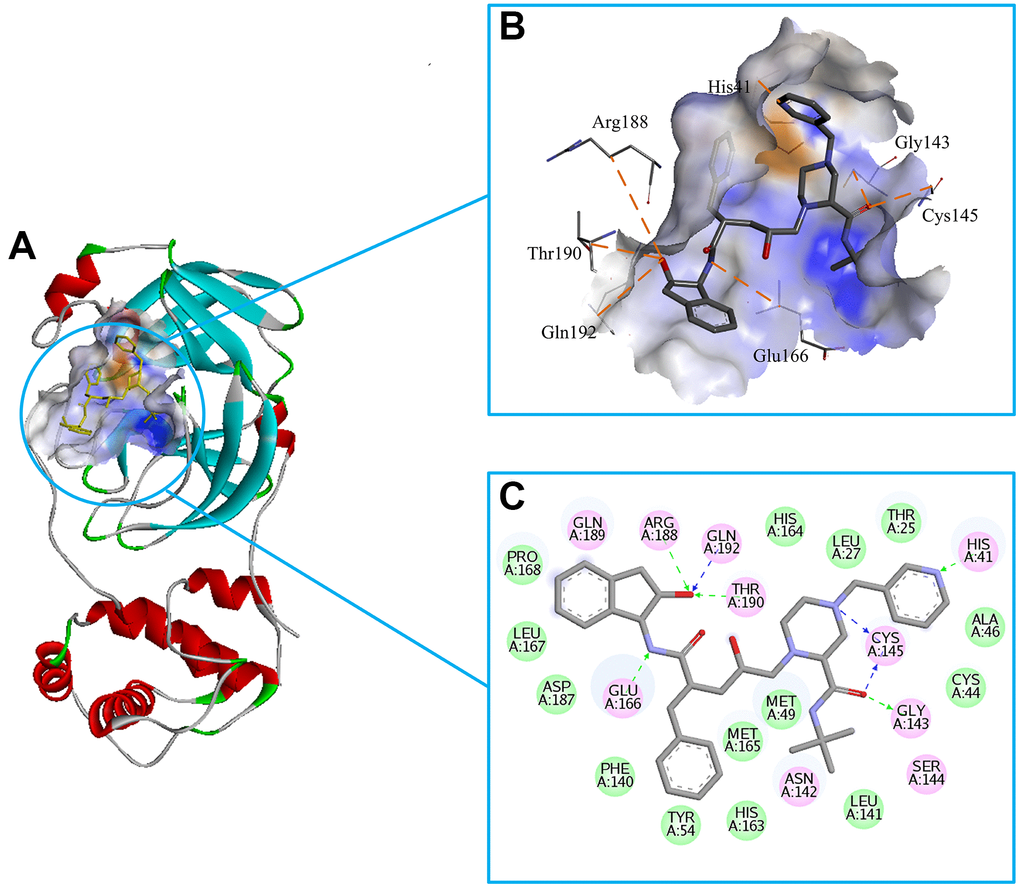
Figure 2. Investigation on the binding pattern of 3CLpro and Indinavir. (A) The location of the binding pocket between 3CLpro and Indinavir. (B) The enlarged view of the binding pocket of 3CLpro and Indinavir. (C) The interaction 2D diagram between 3CLpro and Indinavir. The green rectangles represent VDW interaction. The pink rectangles represent charge and H-bond interactions. Here, the H-bond interactions with the main residues are indicated by the green dashed arrow pointing to the electron donor, and the H-bond interactions with the side chain residues are indicated by the blue dashed arrow pointing to the electron donor.
The calculation of binding free energy
To better explore how ligand affected the stability and interaction of protein-ligand complexes, the binding free energies of protein-ligand complexes were calculated using the MM/PBSA algorithm. binding free energy was described by four characteristic terms, including VDW interaction energy, electrostatic energy, polar solvation free energy and non-polar solvation free energy. VDW, electrostatic interaction and nonpolar solvation energy had negative contribution to the total interaction energy, while only polar solvation energy had a positive contribution to the total free binding energy, which meant that VDW, electrostatic interaction and non-polar solvation energy were beneficial to enhance the stability of the complex systems. According to Table 2, it could be seen that the binding free energy between 3CLpro protein and Indinavir was -239.796 kJ/mol, indicating that they had high binding affinity.
Table 2. The binding free energy of 3CLpro and Indinavir.
| Complex | Van der Waal (kJ/mol) | Electrostatic (kJ/mol) | Polar solvation (kJ/mol) | Non-polar solvation (kJ/mol) | Binding energy (kJ/mol) |
| 3CLpro-Indinavir | -128.153 | -173.386 | 89.208 | -27.465 | -239.796 |
In order to find out the contribution of different residues to the interaction between the protein-ligand complex, the free energy decomposition method was used to decompose the interaction energy into single residue, and further explored the differences in the sources of binding affinity between Indinavir and 3CLpro protein. The result of free energy decomposition was shown in Figure 3. Among them, the first 10 main residues in the 3CLpro protein that had higher binding free energies with the Indinavir were His41, Ser46, Met49, Leu141, Gly143, Cys145, Met165, Glu166, Arg188 and Gln192, respectively. These main residues were mainly distributed in regions I and II, which could help enhance the binding affinity between the 3CLpro protein and Indinavir.
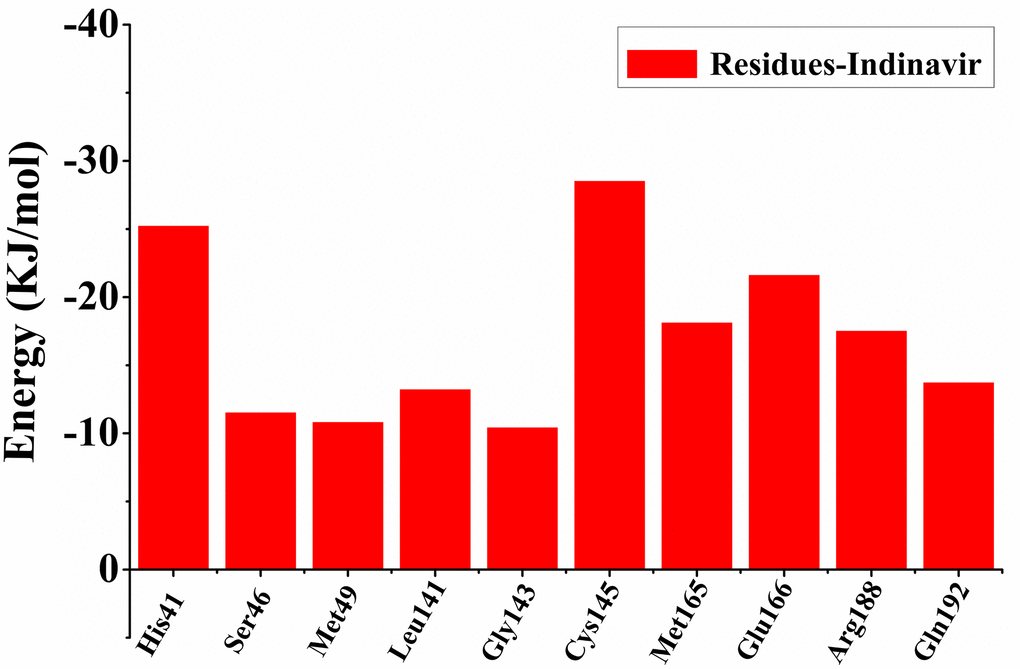
Figure 3. The first 10 main residues in 3CLpro that contribute to the binding free energy of 3CLpro and Indinavir. The ordinate indicates the value of binding free energy, and the abscissa indicates the residue.
Research on stability and flexibility of simulation system
In order to explore the relevant information of protein systems over time, 100 ns molecular dynamics simulations of 3CLpro system (without ligand) and 3CLpro-Indinavir complex system were carried out. The RMSD was used to evaluate the difference of the protein main chain from its initial structural conformation to its final position. Through RMSD analysis, the conformational stability of the system during MD simulation could be evaluated. The fluctuation amplitude of the RMSD curve was inversely related to the stability of the protein, that was, the smaller the fluctuation, the more stable the system. Figure 4A showed the RMSD curves of the skeleton atoms in the 3Clpro system (black curve) and 3CLpro-Indinavir complex system (blue curve). It could be found from the Figure 3 that these two systems tended to converge around 5 ns and reach equilibrium afterwards. The average RMSD values in the 3CLpro and 3CLpro-Indinavir systems were 0.2712 nm and 0.2205 nm, respectively. Since the simulation trajectories of the two systems were stable within 5-100 ns, the simulation trajectories of the last 95 ns were intercepted for later analysis.
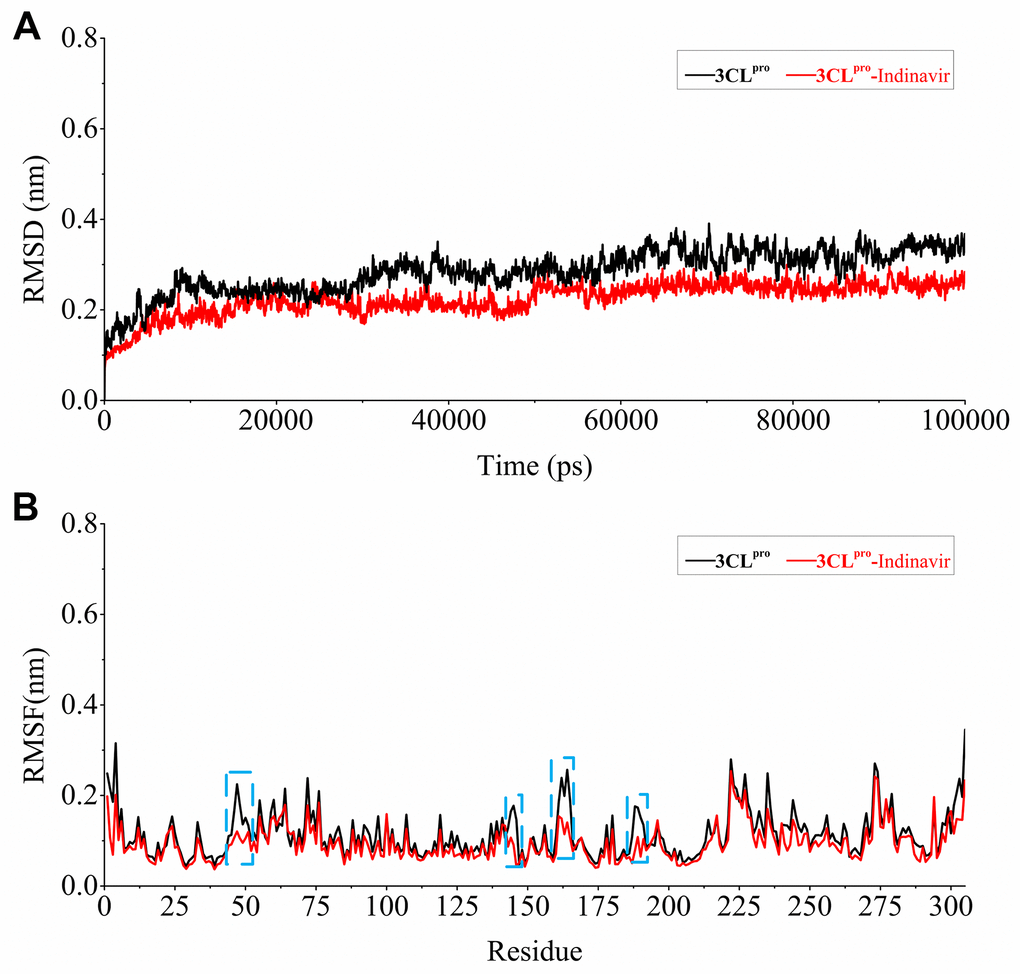
Figure 4. Stability evaluation of 3CLpro system and 3CLpro-Indinavir system. (A) RMSD of all main chain atoms of 3CLpro system and 3CLpro-Indinavir system. (B) RMSF of side chain atoms of 3CLpro system and 3CLpro-Indinavir system. The black line indicates the result of the 3CLpro system, and the red line indicates the result of the 3CLpro-Indinavir system. In addition, these regions (residues Glu47-Tyr54, Phe140-Cys145, His163-Pro168 and Val186-Thr190) are highlighted with blue dotted box.
Residues were protein components that determined its conformational characteristics. This ligand-residue interaction at the active site might induce conformational changes in protein structure and change its function. More specifically, the conformational change was the result of ligand-induced movement during ligand binding. Understanding ligand-induced conformational changes in protein structure was essential for rational drug design based on structure. The RMSF was a measure of the average atomic mobility of skeleton atoms (N, Cα, and C) during MD simulations and was used to assess the flexibility of the side chains of residues [13]. In order to explore the impact of each residue on protein flexibility, the RMSF fluctuation value was calculated, as shown in Figure 4B. The overall RMSF of the 3CLpro and 3CLpro-Indinavir systems showed similar fluctuation trends, and the average values of the RMSF curves were about 0.1382 nm and 0.1266 nm, respectively. It was worth noting that in these regions (residues Glu47-Tyr54, Phe140-Cys145, His163-Pro168 and Val186-Thr190, marked with blue dotted boxes), compared with the 3CLpro system, the 3CLpro-Indinavir system showed significantly reduced fluctuations. Moreover, since these four regions were mainly located in the catalytic binding region between protein domains I and II, this indicated that the 3CLpro protein could exhibit lower flexibility and contribute to stabilize the protein conformation after binging with Indinavir.
Investigation on the conformational transitions and dynamic domain motions of the 3CLpro system and the 3CLpro-Indinavir complex system
In order to explore the effect of the Indinavir on the conformational state of 3CLpro protein, the last 95 ns of MD simulation trajectories were used to perform PCA on the Cα atoms from the 3CLpro and 3CLpro-Indinavir systems. By projecting the simulated trajectories of the protein systems into the two-dimensional subspace spanned by the first three eigenvectors (PC1, PC2 and PC3), the PCA scatter plots of the 3CLpro and 3Clpro-Indinavir systems were obtained to explore the conformational transformation of the systems. As shown in Figure 5, the top 20 PCs of the 3CLpro system and 3CLpro-Indinavir system accounted for 83.1% and 77.8% of the total change, respectively, indicating that compared with the 3CLpro system, the 3CLpro-Indinavir system had a smaller phase space and a lower performance flexibility. In the 3CLpro system, PC1 and PC2 contributed 36.3% and 13.9% to the variance, respectively, while other PCs contributed no more than 9.0%. In the 3CLpro-Indinavir system, PC1 and PC2 contributed 30.3% and 12.0% to the variance, while other PCs also contributed no more than 9.0%. This showed that the first two eigenvectors (PC1 and PC2) could largely reflect the transformation of the conformational state to a large extent, so we projected along the direction of the first two main components (PC1 and PC2) to generate PCA scatter plots (Figure 5). As shown in the figure, the PCA scatter diagram showed the conformational state of the two systems in the subspace, where the red dot represented the stable conformational state, the blue dot represented the unstable conformational state, and the intermediate state between the two conformations was represented by the white dots. The protein system periodically jumped between the two conformational states (blue and red). Comparing the scatter plots of the two systems, it could be seen that the conformational states of the protein after binding with the Indinavir had changed significantly. Compared with the irregular distribution of red and blue dots in the 3CLpro system, the red and blue dots in the 3CLpro-Indinavir complex system were evenly distributed along the diagonal on both sides of the diagonal, indicating that the Indinavir contributed to stabilize the conformational state of 3CLpro protein, which was consistent with the RMSF result.
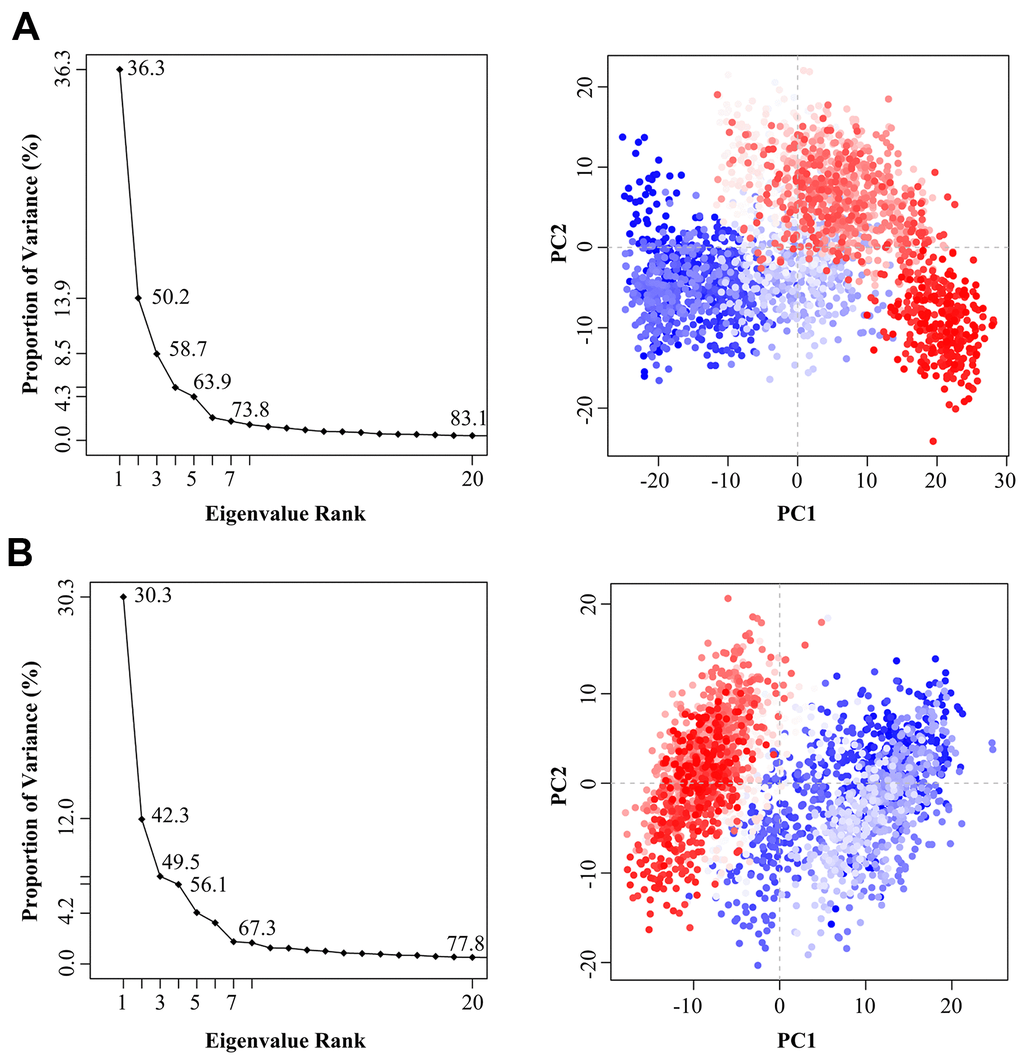
Figure 5. The variance contribution of the principal components and the PCA scatter plots generated along the projection of the first two eigenvectors (PC1 and PC2) in the space of the 3CLpro (A) and 3CLpro-Indinavir (B) systems, respectively.
In order to explore the effect of the Indinavir on the conformational motions of 3CLpro protein, DCCM analyses were performed on all Cα atoms in the 3CLpro system and the 3CLpro-Indinavir complex system by extracting the last 95 ns simulated trajectories. The 2D diagrams of DCCM showed the correlated motions between residues during the whole simulation process (Figure 6). DCCM showed the overall correlation, which ranged from -1.0 to 1.0 (from dark purple to dark blue). Different colors were used to define different degrees of correlation between residues, and the darker the color, the stronger the correlation. The positive correlation (from 0 to 1) meant that the residues moved in the same direction, while the negative correlation (from -1 to 0) meant that the residues moved in the opposite direction. Comparing the DCCM diagrams of the two systems, it could be found that the correlated motions of the two systems were significantly different. Compared with the 3CLpro system, the positive correlation motions of the entire 3CLpro-Indinavir system had not change much, but the negative correlation motions were significantly reduced. Especially in some important areas (marked with black dashed boxes), the correlated motions of the protein after binding with ligand would change significantly. Compared with the 3CLpro system, in the 3CLpro-Indinavir system, the region (residues Leu250-Glu270) and region (residues Asn142-Met162), region (residues Gly215-Met235) and region (residues Gly120-Cys145), and region (residues Phe230-Cys265) and region (residues Leu30-Leu50) had significantly reduced negative correlation motions, which could lead to reduced flexibility of residues. Therefore, the reduction of negative correlation motions might be conducive to stabilizing the conformational state of protein domains I and II, so that the catalytically active site of the protein tended to be conserved, thereby inactivating the protein.
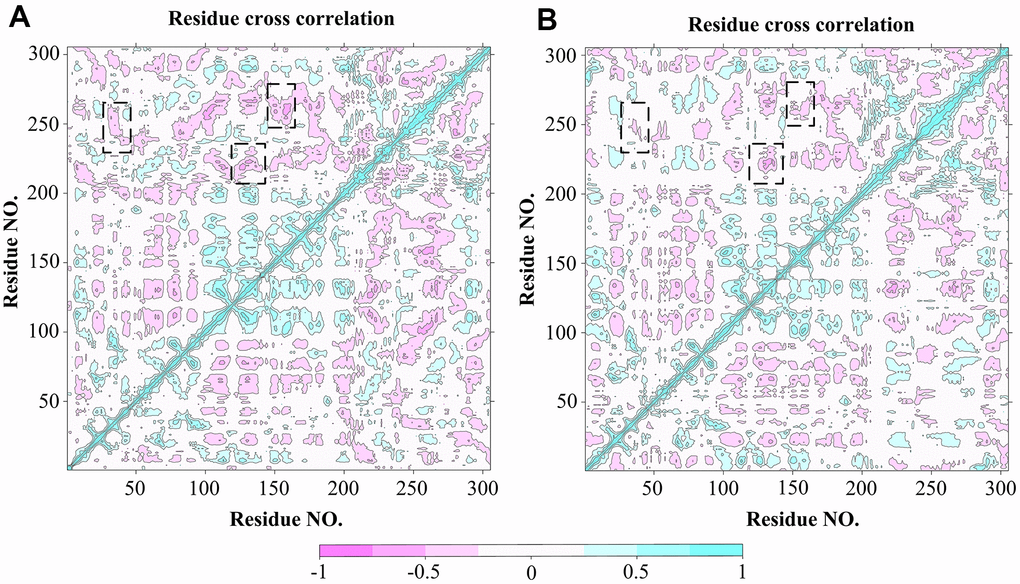
Figure 6. DCCM analysis of Cα atoms for 3CLpro system (A) and 3CLpro-Indinavir system (B), respectively. The red regions indicate negative correlation, and the blue regions indicate positive correlation. The darker the color, the stronger the correlation. Regions with significant differences in correlated motions have been marked with black dashed boxes.
The investigation of differences in the residue interactions between 3CLpro system and 3CLpro-Indinavir system
In order to further explore the structure and function of 3CLpro protein after binding with the Indinavir, RINs were performed on the 3CLpro system and 3CLpro-Indinavir system to analyze the difference in the interaction between key residues. We focused on observing the differences in the residue-residue interactions around the catalytic dyads His41-Cys145 between the two systems. Figure 7 showed the RIN diagrams of the residue-residue interactions around the catalytic dyads His41-Cys145 of the two systems. According to the RIN results, the residue-residue interactions in the 3CLpro protein would change significantly after binding with the Indinavir, which was mainly manifested in the significant increase in the H-bond interactions and VDW interactions formed between residues and residues. In the 3CLpro system, Residue His41 formed an H-bond interaction with Cys44, and formed the VDW interactions with Tyr54 and His164. Residue Cys145 formed H-bond interaction with His164, and formed the VDW interactions with Leu27 and Asn28. In the 3CLpro-Indinavir system, residue His41 formed H-bond interactions with Cys44, Cys145 and His164, and formed VDW interactions with Met49 and Tyr54. Residue 145 formed the VDW interaction with Leu27, and formed H-bond interactions and VDW interactions with Asn28 and His164. It was worth noting that in the 3CLpro-Indinavir system, residue Cys145 formed new H bond interactions with Asn28 and His41, and formed new VDW interactions with His164. Residue His41 formed a new VDW interaction with Met49, and the interaction with His164 was transformed from VDW interaction to H-bond interaction. It could be clearly found that the interactions between the catalytic dyads His41-Cys145 and its surrounding residues had increased significantly. Therefore, the combination of 3CLpro and Indinavir could significantly increase the residue-residue interaction at the catalytic site, which was conducive to stabilizing the protein conformation, and further revealed the underlying reason for the inactivation of 3CLpro protein after binding with the Indinavir.
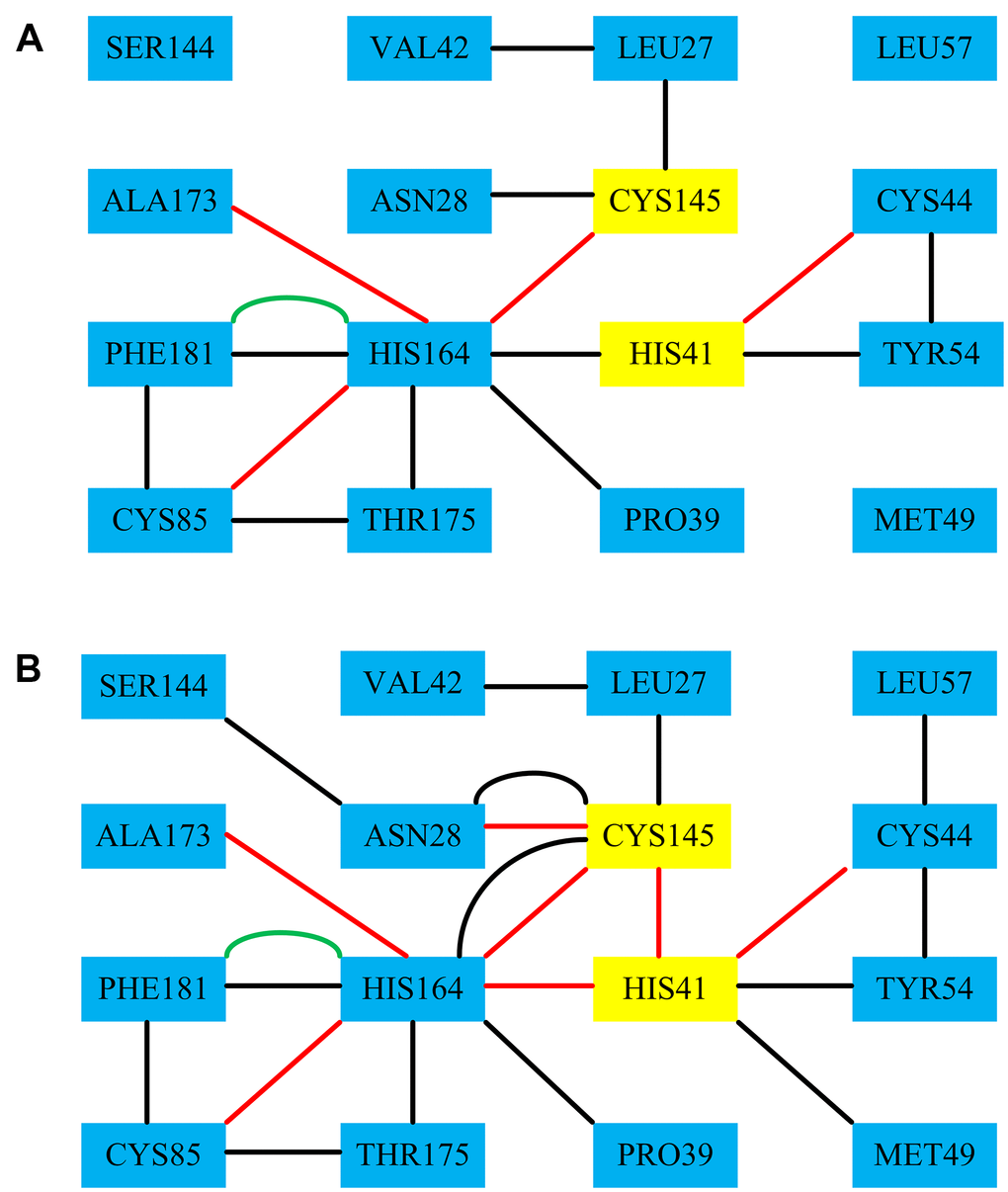
Figure 7. The RINs results of the residues-residues around the catalytic dyads His41-Cys145 of the 3CLpro system and the 3CLpro-Indinavir system, respectively. Among them, the red line represents the H-bond interaction, the black line represents the VDW interaction, and the green line represents the Pi-Pi interaction.
The analysis on the H-bond occupancy
The residue-residue H-bond interaction was considered to be the most important factor affecting the conformational state of the protein. In order to verify whether the H-bond interaction shown on RINs in the 3CLpro system and 3CLpro-Indinavir complex system was formed stably during the MD simulations, the H-bond occupancy was calculated. When the H-bond occupancy exceeded 50%, it indicated that the H-bond could be formed stably [14]. Table 3 showed the calculation results of the H-bond occupancy in the 3CLpro system and the 3CLpro-Indinavir system during the 100 ns simulation process. In the 3CLpro system, His41-Cys44, Cys145-Cys164, His164 and Cys85 and His164-Ala173 could form stable H-bond interactions, and the values of H-bond occupancy were 82.56%, 84.31%, 80.46% and 75.88%, respectively. In the 3CLpro-Indinavir system, His41-Cys44, His41-Cys145, His41-His164, Cys145-Asn28, Cys145-Cys164, His164-Cys85, His164-Cys85 and His164-Ala173 could all form stable H-bond interactions, and values of their H-bond occupancy were 92.33%, 98.28%, 89.37%, 87.64%, 95.98%, 88.19% and 91.49%, respectively. It was worth noting that in the 3CLpro-Indinavir system, His41-Cys145, His41-His164 and Cys145-Asn28 formed new H-bond interactions, and values of their H-bond occupancy exceeded 50%, indicating that these H-bond interactions were very stable, which further supported the results of RINs.
Table 3. The occupancy of H-bond interactions during the simulations of 3CLpro system and 3CLpro-Indinavir system, respectively.
| Interaction type | Residue 1 | Residue 2 | Occupancy (%) (3CLpro) | Occupancy (%) (3CLpro- Indinavir) |
| H bond | His41 | Cys44 | 82.56 | 92.33 |
| H bond | His41 | Cys145 | 11.24 | 98.28 |
| H bond | His41 | His164 | 20.66 | 89.37 |
| H bond | Cys145 | Asn28 | 21.02 | 87.64 |
| H bond | Cys145 | Cys164 | 84.31 | 95.98 |
| H bond | His164 | Cys85 | 80.46 | 88.19 |
| H bond | His164 | Ala173 | 75.88 | 91.49 |
Discussion
COVID-19 continues to ravage the world, bringing a heavy burden to people all over the world, so we are eager to find a solution. 3CLpro protein had become an attractive target for the treatment of SARS-COV-2 due to its key role in the virus replication cycle. In this study, we used the 3CLpro protein as a target to conduct high-throughput virtual screening of FDA-approved drugs to find potential drugs against COVID-19. According to the level of the docking score, 10 potential drugs that could inhibit SARS-COV-2 were identified. Among them, The Indinavir and 3CLpro protein had more advantages in docking result and binding affinity. Studies by other people had also found that Indinavir and other FDA-approved drugs had potential advantages in the process of binding to 3CLpro protein, which was consistent with our research, which indicated that drugs such as Indinavir could be used as the inhibitor of 3CLpro protein in subsequent study [15, 16]. Unfortunately, their research lacked in-depth exploration of its binding mechanism, and the conformational differences of the 3CLpro protein after binding to Indinavir and other drugs were unclear.
Therefore, in order to explore the conformational differences of the 3CLpro protein after binding with Indinavir, the 100 ns molecular dynamics simulations were performed on the 3CLpro system and the 3CLpro-Indinavir complex system for the first time in this study, respectively, and the post-dynamic analyses of the simulated trajectories of the two systems was performed. Firstly, RMSD and RMSF analyses showed that both systems reached equilibrium around 5 ns, and the 3CLpro-Indinavir system had higher stability. Then, through PCA and DCCM, the conformational states of these two systems was explored, and it was found that the 3CLpro-Indinavir system had a smaller phase space and the negatively correlated movement of the system was significantly reduced. Finally, the RIN and H-bond occupancy were analyzed, and it was found that the residue-residue interactions in 3CLpro protein increased significantly after binding with Indinavir, especially the catalytic dyads His41-Cys145 could form multiple stable H-bond and VDW interactions with surrounding residues., which revealed the deeper reason why Indinavir inhibited 3CLpro protein. In short, although our research is based on bioinformatics, its effectiveness needs to be verified by subsequent in vitro tests. Through this research, we obtained some FDA-approved drugs that might have potential inhibitory effects on 3CLpro protein, and further clarified the conformational differences of 3CLpro protein after binding with Indinavir, which provided valuable information for subsequent drug development and optimization. We hope to provide more new clues against COVID-19.
Materials and Methods
System preparation
The crystal structure of 3CLpro protein (PDB ID: 6LU7) was downloaded from the Protein Data Bank (PDB) [9]. The protein was prepared through the " Prepare Protein " module in Discovery Studio (DS) v3.5 software, including removing water, adding the hydrogen atoms, assigning bond order, treating metals, treating disulfides, and inserting missing residues. In addition, ligands were prepared by DS v3.5 "prepare ligand" module, which included maintaining ionization, desalting, generating tautomer and converting the 2D conformation to the 3D conformation.
High-throughput virtual screening based on molecular docking
Discovery Studio V3.5 software was used for high-throughput virtual screening. We downloaded all FDA-approved ligand structure models from the ZINC database. These compounds were prepared before docking screening.
The model of the interaction between protein and ligand was obtained by executing the "CDOCKER" module, which was based on the docking method of CHARMM. The "Define and Edit Binding Site" tool was used to define the binding pocket and select all amino acid residues within 5 Å around the ligand [17]. The key amino acid residues at the 3CLpro protein binding site include His41, Met49, Tyr54, Phe140, Leu141, Asn142, Cys145, His163, Met165, Glu166, Leu167, Phe185, Asn187, and Gln192 [11]. The compounds were docked into the receptor protein and ranked according to the docking score, where the -CDOKER_ENERGY value indicated the docking score, and the higher the docking score, the better the compound bound to the protein.
MD simulation
MD simulation was performed in the GROMOS96 43a1 force field using the GROMACS 4.5.5 software package [18]. Firstly, through the generated "topology" file, the non-bonding parameters (atom types and charges) and bonding parameters (bonds, angles, and dihedrals) were defined in the simulation. Then, all models were simulated in a closed dodecahedral box filled with explicit single-point charge (SPC) water molecules, with a minimum distance of 1 nm between the protein surface and the box boundary. To neutralize the system, the appropriate amount of counter ions was added to the system. Subsequently, the steepest descent method was used to minimize the energy of each system to 1000 kJ·mol-1/nm to ensure that there were no spatial collisions and inappropriate geometry inside the system. The position-restrained dynamics simulation (namely NVT and NPT) better balanced the solvent and ions around the protein model. Here, N was the number of particles, P was the system pressure, V was the volume, and T was the temperature. During the 100 ps NVT simulation, all systems were heated from 0 K to 300 K and stabilized at 300 K with the help of the thermostat. During the 100 ps NPT simulation, the constant pressure device was used to remove the constraints and keep the constant pressure at 1 bar. All bonds in the system were constrained by the LINCS algorithm [19]. Finally, 100 ns MD simulation was performed.
Principal component analysis (PCA)
As a statistical method, PCA was widely used to reduce the dimensionality of data obtained from molecular dynamics simulations, and could be used to extract dominant modes in molecular motion [20, 21]. The first two eigenvectors (PC1 and PC2) of the maximum motions were obtained by projection to identify the motion of the protein. Each main movement was characterized by an eigenvector and an eigenvalue. The eigenvector meant the direction of motion, and the eigenvalue meant the contribution of a specific component to the overall motion of the complex [22, 23]. The ensemble formula used to generate the elements Cij with coordinates i and j as the covariance matrix was defined as follows:
Cij = <(ri − <ri>) (rj − <rj>)>
Where, ri meant the Cartesian coordinates of the ith Ca atom and rj meant the Cartesian coordinates of the jth Cα atom; and the Angular brackets “<>” meant the time average of all relevant configurations during the entire simulation; and i and j meant the number of Cα atom. The dynamic motion of the atoms in the system was calculated from the simulation trajectories to analyze the conformational differences between the system during the simulation [24, 25]. This analysis was performed using Bio3D library [26].
Dynamic cross-correlation map (DCCM)
Cross-correlation was a 3D matrix representation that could display time-related information between protein residues [27]. In biomolecules, the relative movement of Cα atoms helped to identify the binding area. DCCM was used to detect the time-dependent motions of all Cα atom pairs, and was calculated according to the following formula: [28, 29]
Cij = <Δri • Δrj>/(<Δri2><Δrj2>)1/2
In the formula, i and j indicated the i-th and j-th atoms, respectively. The Δi and Δj indicated the displacement vector corresponding to i-th and j-th atoms. And the <.......> indicated the ensemble average. The cross-correlation coefficient Cij varied from -1 to 1 [30, 31]. A positive value of Cij indicated that the motion of atoms was interrelated and in the same direction, and a negative value of Cij indicated that the residues were inversely correlated [32].
Residue interaction network (RIN)
The residue interaction network (RIN) was a reliable tool for identifying functional residues and analyzing changes in the structure of the interaction network between proteins and ligands [33, 34]. RIN analysis had been widely used to analyze a wide range of biological processes, including mutational effects, protein folding, intra protein domain-domain communication and catalytic activity [35]. RIN was visualized by Cytoscape software [36]. The average structure in the simulated trajectory was extracted to construct the RIN defined as the contact network. In the 2D diagram of RIN, nodes represent residues, and the lines between them represent the interactions between residues and residues, including H-bond interactions and VDW interactions [37].
Binding free energy
The molecular mechanics Poisson Boltzmann surface area (MM-PBSA) method was used to calculate the binding free energy between residues and ligand [38]. The binding free energy could be calculated by the following formula:
∆Gbind = Gcomplex – Greceptor – Gligand
Here, Gcomplex, Greceptor and Gligand were the free energies of complex, receptor and ligand, respectively.
∆G = ∆Egas + ∆Gsol − T∆S
Here, the binding free energy (∆G) contained gas phase free energy (ΔEgas), solvation free energy (∆Gsol) and conformational entropy (T∆S). The free energy (∆Egas) consisted of three parts, including internal energy (∆Eint), electrostatic interaction (∆Eele) and van der Waals force (∆Evdw). Solvation free energy (∆Gsol) includes electrostatic solvation free energy (∆Gpolar) and nonpolar solvation free energy (∆Gnonpolar) [23, 39].
Author Contributions
Wen-Shan Liu, Han-Gao Li and Jia-Qiu Li conceived and designed the experiment. Chuan-Hua Ding, Hai-Xia Zhang and Rui-Rui Wang performed the experiments and analyzed the data. All authors contributed to write and edited the paper.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This study was supported by the Project of Shandong Provincial Natural Science Foundation of China (Contract grant numbers: ZR2019BH009).
References
- 1. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579:270–73. https://doi.org/10.1038/s41586-020-2012-7 [PubMed]
- 2. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, et al, and China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020; 382:727–33. https://doi.org/10.1056/NEJMoa2001017 [PubMed]
- 3. Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun. 2020; 109:102433. https://doi.org/10.1016/j.jaut.2020.102433 [PubMed]
- 4. Wei H, Yin H, Huang M, Guo Z. The 2019 novel cornoavirus pneumonia with onset of oculomotor nerve palsy: a case study. J Neurol. 2020; 267:1550–53. https://doi.org/10.1007/s00415-020-09773-9 [PubMed]
- 5. Tahir Ul Qamar M, Alqahtani SM, Alamri MA, Chen LL. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020; 10:313–19. https://doi.org/10.1016/j.jpha.2020.03.009 [PubMed]
- 6. Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, Zhong W, Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020; 63:457–60. https://doi.org/10.1007/s11427-020-1637-5 [PubMed]
- 7. Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020; 368:409–12. https://doi.org/10.1126/science.abb3405 [PubMed]
- 8. Hegyi A, Ziebuhr J. Conservation of substrate specificities among coronavirus main proteases. J Gen Virol. 2002; 83:595–99. https://doi.org/10.1099/0022-1317-83-3-595 [PubMed]
- 9. Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020; 582:289–93. https://doi.org/10.1038/s41586-020-2223-y [PubMed]
- 10. Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002; 21:3213–24. https://doi.org/10.1093/emboj/cdf327 [PubMed]
- 11. Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA. 2003; 100:13190–95. https://doi.org/10.1073/pnas.1835675100 [PubMed]
- 12. Liu WS, Wang RR, Sun YZ, Li WY, Li HL, Liu CL, Ma Y, Wang RL. Exploring the effect of inhibitor AKB-9778 on VE-PTP by molecular docking and molecular dynamics simulation. J Cell Biochem. 2019; 120:17015–29. https://doi.org/10.1002/jcb.28963 [PubMed]
- 13. Pol-Fachin L, Fernandes CL, Verli H. GROMOS96 43a1 performance on the characterization of glycoprotein conformational ensembles through molecular dynamics simulations. Carbohydr Res. 2009; 344:491–500. https://doi.org/10.1016/j.carres.2008.12.025 [PubMed]
- 14. Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2008; 4:116–22. https://doi.org/10.1021/ct700200b [PubMed]
- 15. Hall DC
Jr , Ji HF. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med Infect Dis. 2020; 35:101646. https://doi.org/10.1016/j.tmaid.2020.101646 [PubMed] - 16. Kumar S, Sharma PP, Shankar U, Kumar D, Joshi SK, Pena L, Durvasula R, Kumar A, Kempaiah P, Poonam, Rathi B. Discovery of New Hydroxyethylamine Analogs against 3CLpro Protein Target of SARS-CoV-2: Molecular Docking, Molecular Dynamics Simulation, and Structure-Activity Relationship Studies. J Chem Inf Model. 2020; 60:5754–70. https://doi.org/10.1021/acs.jcim.0c00326 [PubMed]
- 17. David CC, Jacobs DJ. Principal component analysis: a method for determining the essential dynamics of proteins. Methods Mol Biol. 2014; 1084:193–226. https://doi.org/10.1007/978-1-62703-658-0_11 [PubMed]
- 18. Li HL, Ma Y, Zheng CJ, Jin WY, Liu WS, Wang RL. Exploring the effect of D61G mutation on SHP2 cause gain of function activity by a molecular dynamics study. J Biomol Struct Dyn. 2018; 36:3856–68. https://doi.org/10.1080/07391102.2017.1402709 [PubMed]
- 19. Vaccaro L, Koronakis V, Sansom MS. Flexibility in a drug transport accessory protein: molecular dynamics simulations of MexA. Biophys J. 2006; 91:558–64. https://doi.org/10.1529/biophysj.105.080010 [PubMed]
- 20. Kan W, Fang F, Chen L, Wang R, Deng Q. Influence of the R823W mutation on the interaction of the ANKS6-ANKS3: insights from molecular dynamics simulation and free energy analysis. J Biomol Struct Dyn. 2016; 34:1113–22. https://doi.org/10.1080/07391102.2015.1071281 [PubMed]
- 21. Zhou Y, Zhang N, Chen W, Zhao L, Zhong R. Underlying mechanisms of cyclic peptide inhibitors interrupting the interaction of CK2α/CK2β: comparative molecular dynamics simulation studies. Phys Chem Chem Phys. 2016; 18:9202–10. https://doi.org/10.1039/c5cp06276d [PubMed]
- 22. Fakhar Z, Govender T, Maguire GE, Lamichhane G, Walker RC, Kruger HG, Honarparvar B. Differential flap dynamics in l,d-transpeptidase2 from mycobacterium tuberculosis revealed by molecular dynamics. Mol Biosyst. 2017; 13:1223–34. https://doi.org/10.1039/c7mb00110j [PubMed]
- 23. Grant BJ, Rodrigues AP, ElSawy KM, McCammon JA, Caves LS. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006; 22:2695–96. https://doi.org/10.1093/bioinformatics/btl461 [PubMed]
- 24. Arnold GE, Ornstein RL. Molecular dynamics study of time-correlated protein domain motions and molecular flexibility: cytochrome P450BM-3. Biophys J. 1997; 73:1147–59. https://doi.org/10.1016/S0006-3495(97)78147-5 [PubMed]
- 25. Ichiye T, Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991; 11:205–17. https://doi.org/10.1002/prot.340110305 [PubMed]
- 26. Kumar P, Ghosh Sachan S, Poddar R. Mutational analysis of microbial hydroxycinnamoyl-CoA hydratase-lyase (HCHL) towards enhancement of binding affinity: a computational approach. J Mol Graph Model. 2017; 77:94–105. https://doi.org/10.1016/j.jmgm.2017.08.014 [PubMed]
- 27. Wan H, Hu JP, Tian XH, Chang S. Molecular dynamics simulations of wild type and mutants of human complement receptor 2 complexed with C3d. Phys Chem Chem Phys. 2013; 15:1241–51. https://doi.org/10.1039/c2cp41388d [PubMed]
- 28. Li X, Wang X, Tian Z, Zhao H, Liang D, Li W, Qiu Y, Lu S. Structural basis of valmerins as dual inhibitors of GSK3β/CDK5. J Mol Model. 2014; 20:2407. https://doi.org/10.1007/s00894-014-2407-1 [PubMed]
- 29. Xu L, Kong R, Zhu J, Sun H, Chang S. Unraveling the conformational determinants of LARP7 and 7SK small nuclear RNA by theoretical approaches. Mol Biosyst. 2016; 12:2613–21. https://doi.org/10.1039/c6mb00252h [PubMed]
- 30. Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, Venger I, Pietrokovski S. Network analysis of protein structures identifies functional residues. J Mol Biol. 2004; 344:1135–46. https://doi.org/10.1016/j.jmb.2004.10.055 [PubMed]
- 31. Martin AJ, Vidotto M, Boscariol F, Di Domenico T, Walsh I, Tosatto SC. RING: networking interacting residues, evolutionary information and energetics in protein structures. Bioinformatics. 2011; 27:2003–05. https://doi.org/10.1093/bioinformatics/btr191 [PubMed]
- 32. Hu G, Yan W, Zhou J, Shen B. Residue interaction network analysis of dronpa and a DNA clamp. J Theor Biol. 2014; 348:55–64. https://doi.org/10.1016/j.jtbi.2014.01.023 [PubMed]
- 33. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–504. https://doi.org/10.1101/gr.1239303 [PubMed]
- 34. Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999; 286:509–12. https://doi.org/10.1126/science.286.5439.509 [PubMed]
- 35. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000; 33:889–97. https://doi.org/10.1021/ar000033j [PubMed]
- 36. Zhang YJ, Ding JN, Zhong H, Han JG. Exploration micromechanism of VP35 IID interaction and recognition dsRNA: a molecular dynamics simulation. Proteins. 2017; 85:1008–23. https://doi.org/10.1002/prot.25269 [PubMed]
- 37. Neves BJ, Braga RC, Melo-Filho CC, Moreira-Filho JT, Muratov EN, Andrade CH. QSAR-based virtual screening: advances and applications in drug discovery. Front Pharmacol. 2018; 9:1275. https://doi.org/10.3389/fphar.2018.01275 [PubMed]
- 38. Ul Haq F, Abro A, Raza S, Liedl KR, Azam SS. Molecular dynamics simulation studies of novel β-lactamase inhibitor. J Mol Graph Model. 2017; 74:143–52. https://doi.org/10.1016/j.jmgm.2017.03.002 [PubMed]
- 39. Möglich A, Joder K, Kiefhaber T. End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc Natl Acad Sci USA. 2006; 103:12394–99. https://doi.org/10.1073/pnas.0604748103 [PubMed]