



Introduction
Melanoma is a highly malignant tumor originating from melanocytes; its prevalence lately shows a global annual increase of about 3%-7% [1]. The prognosis is favorable when the disease is detected at an early stage (stages I and II); however, the prognosis of advanced diseases (stages III and IV) is extremely poor, and the 5-year survival rate on conventional treatment (including chemotherapy and biotherapy) is only 5-10% [2]. Melanoma development is strongly influenced by inflammation and immune cell infiltration [3]. In fact, immune checkpoint inhibitors (ICIs), especially anti-PD-1 and anti-PD-L1 blockers, have changed the traditional treatment mode and achieved unprecedented long-lasting responses in patients with melanoma. However, the effective rate of anti-PD-1 blockade fluctuates between 20% and 40%, and the complete response rate is only about 5% [4]. Therefore, risk stratification of melanoma patients by the combination of immune-related factors and pathological classification is key to predict the prognosis and treatment response.
Long noncoding RNAs (lncRNAs) refer to RNA molecules longer than 200 nucleotides which lack protein coding function. lncRNAs play an extensive regulatory role in different stages of tumor immune response, such as antigen exposure, antigen recognition, immune activation, immune cell infiltration, and tumor clearance [5]. Previous studies showed that a variety of lncRNAs, such as UCA1, DSCAM-AS1 and MIR155HG, can promote melanoma development and are potential therapeutic targets [6–8]. However, the role of immune-related lncRNAs in the prognosis of melanoma is still unclear. In this study, gene expression profiles from the TCGA database were analyzed to establish a relationship between immune-related lncRNAs and the prognosis of melanoma patients followed by construction of a new prognostic model.
Results
Characteristics of patients
A total of 470 melanoma patients were analyzed in this study, including 290 males (61.7%) and 180 females (38.3%), with an average age of 59.2 years (18-90 years). The clinicopathological features taken into consideration consisted of age, gender, T stage, lymph node involvement, distant metastasis and clinical stage (Table 1). We drew a flowchart to illustrate the study design and the type of samples involved and analyzed at each step, where each sample represents one melanoma patient (Figure 1).
Table 1. Baseline characteristics of patients with melanoma.
| Characteristics | N(%) |
| Age | |
| <60 | 241(51.3) |
| ≥60 | 221(47.0) |
| unknown | 8(1.7) |
| Gender | |
| Male | 290(61.7) |
| Female | 180(38.3) |
| AJCC stage | |
| I | 77(16.4) |
| II | 140(29.8) |
| III | 171 (36.4) |
| IV | 23(4.9) |
| unknown | 59(12.5) |
| Tumor | |
| T0 | 23(4.9) |
| T1 | 42(8.9) |
| T2 | 78(16.6) |
| T3 | 90(19.1) |
| T4 | 153(32.6) |
| unknown | 84(17.9) |
| Lymph Node | |
| N0 | 235(50.0) |
| N1 | 74(15.7) |
| N2 | 49(10.4) |
| N3 | 55(11.7) |
| Nx | 57(12.2) |
| Metastasis | |
| M0 | 418(88.9) |
| M1 | 24(5.2) |
| Mx | 28(5.9) |
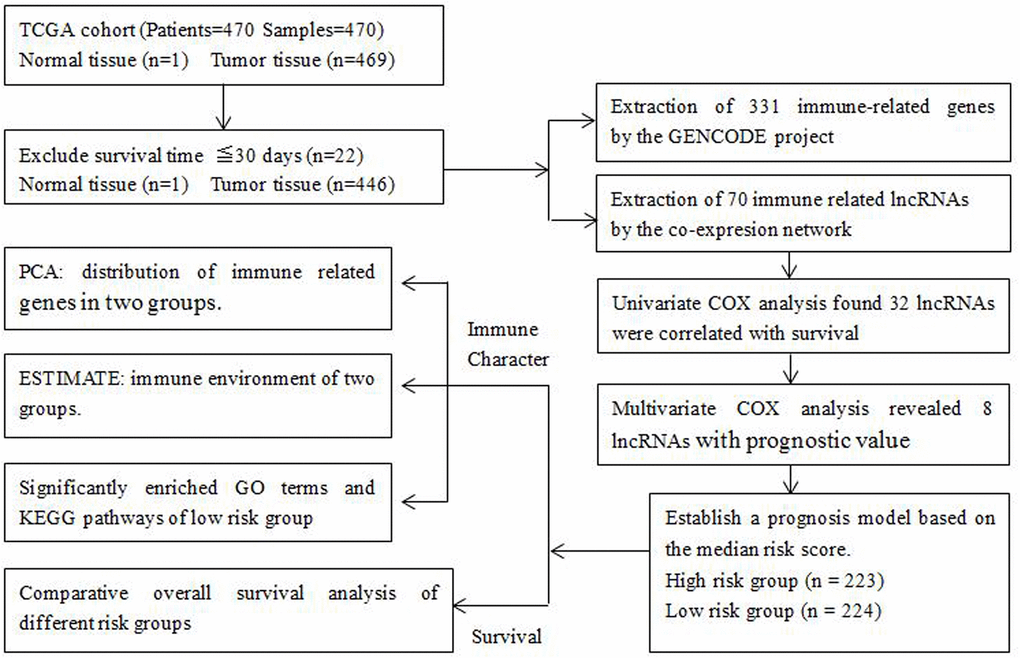
Figure 1. Flowchart of the detailed study design and samples at each stage of the analysis.
Establishment of a prognostic model
Multivariate analysis was performed on the aforementioned 32 immune-related lncRNAs. Eight of 32 lncRNAs were finally identified based on the AIC value, and they were used to construct the prognostic model (Table 3). The risk score of each patient was calculated according to the expression level of each lncRNA and its corresponding regression coefficient. Patients were divided into a high-risk (n = 223) and a low-risk group (n = 224), based on the median risk score. The survival rate was significantly different between two groups (P < 0.001, Figure 2A). The 5-year survival rate of patients in the high- and low-risk groups was 42.7% and 73% respectively. The risk score of each patient and the expression levels of eight immune-related lncRNAs are shown in Figure 2B.
Table 3. Immune-related lncRNAs identified through Cox regression analysis.
| LncRNA | β | HR | P. value |
| AC004847.1 | 0.63 | 1.88 | 0.022 |
| USP30-AS1 | -0.52 | 0.60 | 0.007 |
| U62317.1 | -0.58 | 0.56 | 0.035 |
| MIR205HG | 0.43 | 1.54 | <0.001 |
| AL133371.2 | -0.33 | 0.72 | 0.019 |
| HLA-DQB1-AS1 | -0.25 | 0.78 | 0.048 |
| AL590764.1 | 0.92 | 2.51 | 0.038 |
| AL365361.1 | -0.32 | 0.72 | 0.040 |
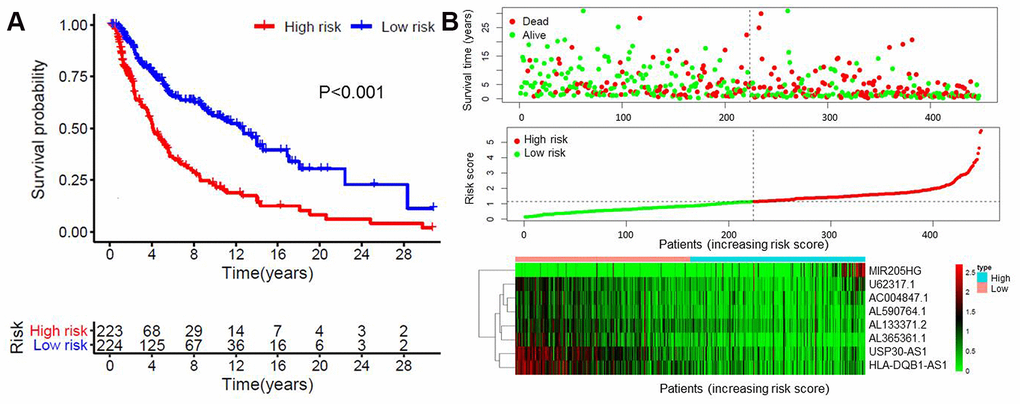
Figure 2. Construction of the eight immune-related lncRNA risk model of melanoma. (A) Kaplan-Meier survival curve of the prognostic model; (B) Risk score distribution, survival status and expression of eight immune-related lncRNAs in high-risk and low-risk groups. Red: high expression; Green: low expression.
Verification and validation of the prognostic model
To further evaluate the ability of the prognostic model in stratification of patients with different TNM stages, we performed survival analyses and found that the model could effectively differentiate the prognosis among patients of stage I (P<0.001), stage II (P=0.023), stage III (P<0.001), and stage IV (P=0.013) (Figure 3A). The area under the ROC curve (AUC) of the prediction model was 0.784 (Figure 3B). In addition, the gene expression profiles of 95 melanoma patients derived from two public datasets (44 samples from GSE98394 and 51 samples from GSE91061) were mined to extract the expression levels of eight lncRNAs and validate the prognostic model using the same algorithm. Of significance, we also observed significant differences in survival between the two risk groups (Figure 3C). The area under the ROC curve (AUC) of the validation dataset was 0.687 (Figure 3D). Our validation analysis demonstrated that this prognostic model can be extended and applied to multiple panels of melanoma cancer patients.
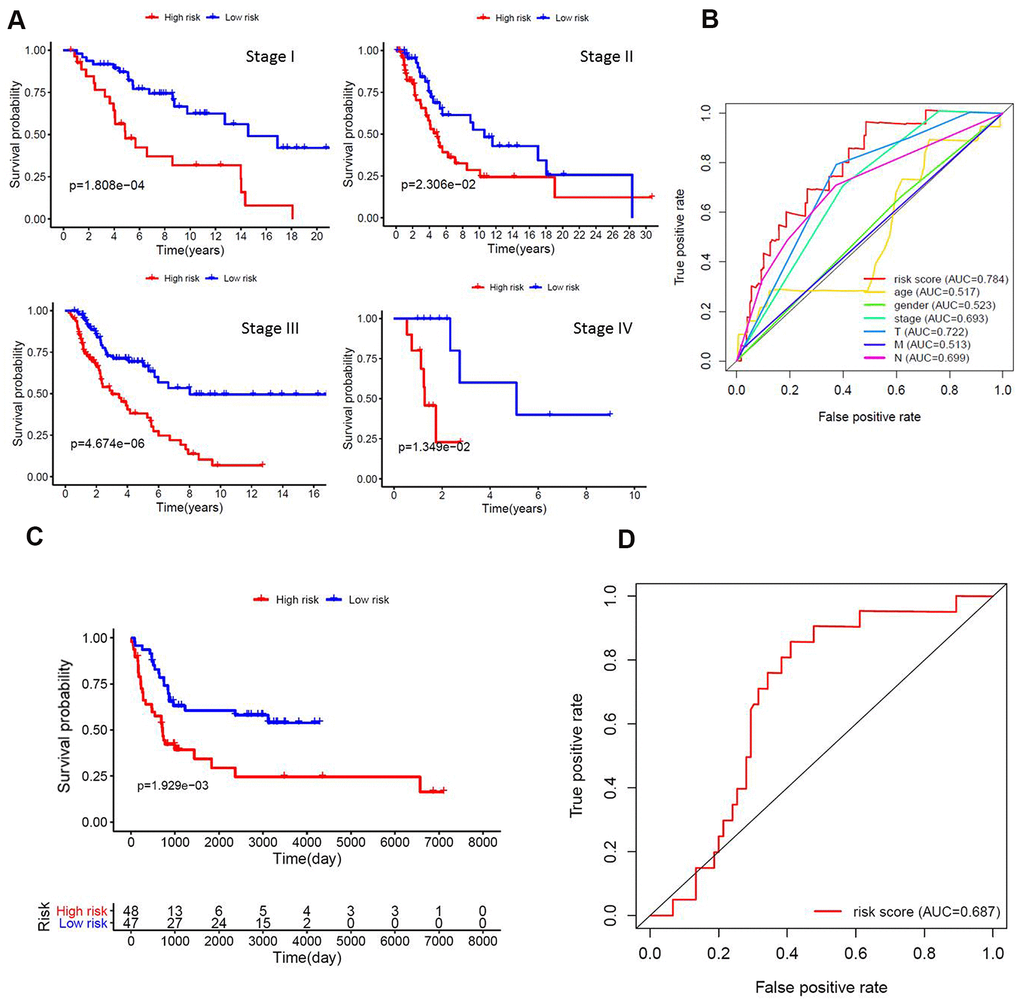
Figure 3. Functional validation of the prognostic model. (A) Kaplan–Meier analysis according to the prognostic model in melanoma patients of different tumor stage; (B) Comparison of ROC curves for prediction of survival by the risk score and other variables (age, gender, stage, tumor, lymph node, metastasis). (C) The gene expression profiles of 95 melanoma patients were mined to extract the expression levels of eight lncRNAs and validate the prognostic model using the same algorithm. We also observed significant differences in survival between the two risk groups; (D) The area under the ROC curve (AUC) of the validation dataset was 0.687.
Immune characteristics of high- and low-risk groups
By principal component analysis (PCA) we found that there were significant differences in the expression profile of immune-related genes within the high- and low-risk groups (Figure 4A, 4B). Functional annotation was further performed by GSEA, revealing that differentially expressed genes between the two groups were enriched in immune system process and immune response pathway signatures (Figure 4C, 4D). After performing the ESTIMATE analysis, we observed that the immune score of the low-risk group was higher than that of the high-risk group (Figure 5A). The low-risk group contained a higher proportion of immune cells and stromal cells; however, the tumor purity is relatively low (P<0.001) (Figure 5B). In the low-risk group, CD8+ T cells were relatively abundant, whereas a lower proportion of M0 macrophages were detected (Figure 5C). Moreover, GO and KEGG enrichment analysis revealed that the biological functions associated the low risk group were mainly concentrated in immune related functions (Table 4).
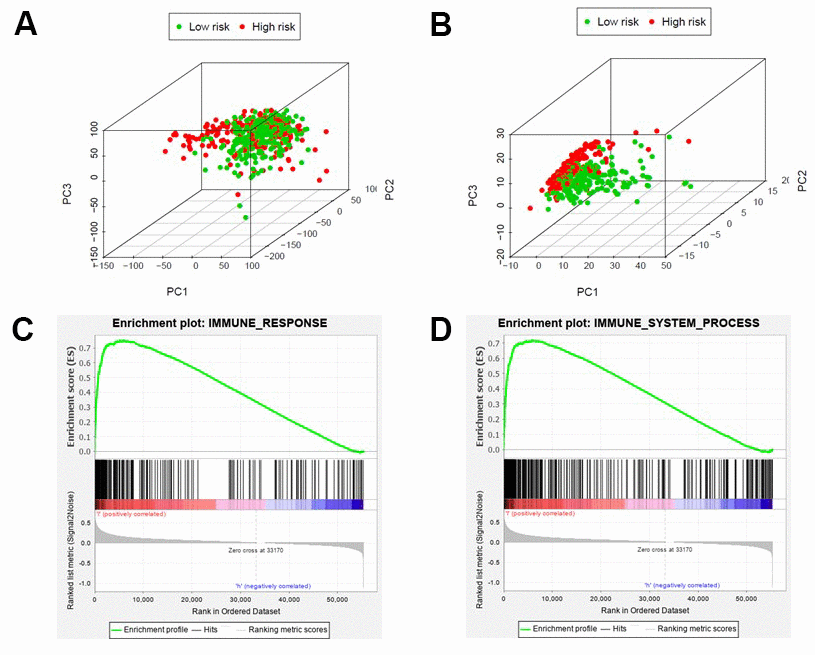
Figure 4. Different immune status in high-risk and low-risk groups. Principal components analysis between high-risk and low-risk groups based on all genes (A) or immune-related genes (B). Enrichment analysis of genes related to immune response (C) or immune system process (D), which shows gene sets enriched in the low-risk group. NSE: Normalized Enrichment Score.
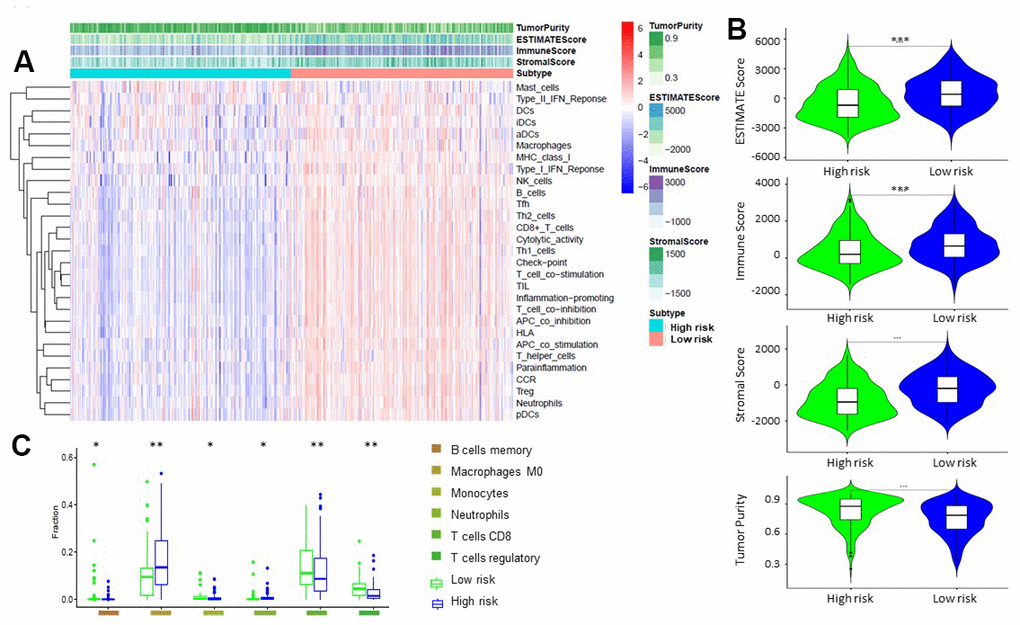
Figure 5. Comparison of ESTIMATE between high-risk and low-risk groups. (A, B) Tumor purity, stromal score, and immune score of different groups were evaluated by ESTIMATE. (C). Comparison of TIICs levels between two groups (ANOVA test).
Table 4. Significantly enriched GO terms and KEGG pathways in the low-risk group.
| GO | ID | Description | NSE | p.adjust | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BP | GO:0045088 | Regulation of innate immune response | 2.65 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BP | GO:0031349 | Positive regulation of defense response | 2.63 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BP | GO:0002218 | Activation of innate immune response | 2.61 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BP | GO:0032088 | Negative regulation of NF-κB transcription factor activity | 2.57 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BP | GO:0009615 | Response to virus | 2.57 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KEGG | hsa05416 | Viral myocarditis | 2.56 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KEGG | hsa04630 | JAK-STAT signaling pathway | 2.46 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KEGG | hsa04650 | Natural killer cell-mediated cytotoxicity | 2.46 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KEGG | hsa04062 | Chemokine signaling pathway | 2.43 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KEGG | hsa04612 | Antigen processing and presentation | 2.43 | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BP, biological process; CC, cellular component; GO, gene ontology; MF, molecular function. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Discussion
The immune system is involved in recognizing and eliminating tumor cells. They can evade this surveillance through immune escape and immunosuppression; indeed, an abnormal immune response is closely associated with tumor development [9]. In recent years, studies have found that lncRNAs play an important regulatory role in the immune response, especially through the following mechanisms: i) they regulate hematopoietic stem cells’ differentiation in the bone marrow into specific mature immune cell populations [10]; ii) they are involved in the peripheral differentiation of dendritic cells, NK cells and innate lymphocytes [11]; iii) they modulate the immune response by controlling the expression of immune-related genes, toll-like receptors (TLRs), and cytokine receptors [12]. iv) they regulate differentiation and activation of T cells and B cells [13]; v) they participate in the activation of autophagy, and other inflammation-associated processes [14]. Immune-related lncRNAs have predictive value for survival and prognosis of a variety of tumors and are potential targets for cancer treatment [15, 16].
In this study, we performed a genome-wide analysis of expression data derived from 470 melanoma patients found in TCGA database, and explored the relationship between immune-related lncRNAs and prognosis of melanoma through survival analysis and Cox regression model. Among the eight immune-related lncRNAs selected to construct the prognostic model, MIR205HG, AC004847.1 and AL590764.1 were poor outcome risk genes, while U62317.1, USP30-AS1, AL133371.2, AL365361.1 and HLA-DQB1-AS1 were protective genes. Previous studies have identified MIR205HG as a potential therapeutic target, as it can promote tumor development and progression by regulating gene transcription [17]; while other lncRNAs have not been characterized previously by relevant clinical or basic studies, and their mechanism of action is still unclear, warranting future investigation.
The incidence and clinical features of melanoma, such as pathological classification, anatomical site and prognosis, are significantly different in different ethnic groups [18]; and the prognosis of patients defined at the same stage is also different. Therefore, a better understanding of the prognostic factors of melanoma is needed. In the proposed model, patients were divided into high- and low-risk groups, and the survival rate between the two groups showed statistically significant differences. AUC (area under the ROC curve) of this prognostic model was 0.784, which was significantly higher than those of the other clinicopathological factors, such as age, gender, T stage, lymph node involvement, distant metastasis and clinical stage. Furthermore, this prognostic model can effectively stratify the risk of melanoma patients belonging to different stages, which indicate that it can be used as a useful complement to the TNM staging system.
Immune checkpoint inhibitors are used as a major therapeutic option for melanoma patients in advanced stage; however, their curative effect depends on the immunogenicity of the tumor. At present, the predictive markers of ICIs in melanoma treatment include the expression level of PD-L1 in tumor cells, the tumor mutation burden (TMB), the presence of tumor infiltrating lymphocytes (TIL), the presence of insertion and deletion (indel) mutations, the number of POLE mutations, specific gut microbiota, etc. [19]. Of course, the characteristics of the tumor microenvironment and of infiltrating lymphocytes play a major role in the process of anti-tumor immunity [20]. In this study, we found that the tumor microenvironment of melanomas in the low-risk group contained larger number of immune cells and stromal cells and lower number of tumor cells. Studies have shown that CD8+T cells, as important effector T cells for tumor elimination, can help to prolong the overall survival of patients and improve the efficacy of immunotherapy [21]. Tumor-associated macrophages (TAM) can express enzymes and cytokines that may be involved in inhibition of the recruitment and activation of T cells, thereby leading to resistance to ICI drugs. Recently, many ongoing clinical trials focus on therapies that inhibit the proliferation or polarization of TAMs to further enhance the anti-tumor immune response [22]. Consistent with previous studies, we found that the low-risk group with higher proportion of CD8+ T cells and lower proportion of M0 macrophages had better prognosis. GO and KEGG enrichment analyses showed that immune-related functions were represented mostly in this group. There were significant differences in terms of expression levels of the immune-related genes in two groups. Of note, the immune score and the PD-L1 expression levels were higher in the low-risk group. Based on the GSEA, the low-risk group contained pathways related to immune response and immune system process. Thus, the low-risk group might be more suitable for immunotherapy as a result of high immunogenicity. Nevertheless, the mechanism is still unclear. Eventually, the response rate to ICIs in different risk groups need to be further evaluated in future studies to validate our findings.
In summary, we analyzed the relationship between immune-related lncRNAs and survival status of melanoma patients and constructed a model to predict the prognosis of patients. There were significant differences in immune status between the high-risk and the low-risk patient group, which may also be applied as a biomarker to evaluate their suitability for immunotherapy. The establishment of this model may thus become convenient for clinicians to choose the most appropriate treatment. We validated this model using two public datasets and their corresponding survival data. Although we observed significant statistical difference of survival outcomes between the high- and low-risk groups, the relatively low frequency of melanoma cancer patients limits the opportunities of validation using additional cohorts. The application value of this proof-of-concept study could be further validated by multi-centric clinical studies including high number of patients.
Materials and Methods
Data collection
The gene expression profile and corresponding prognosis information of 470 patients with melanoma derived tumor and normal tissues were downloaded from the TCGA database (The Cancer Genome Atlas, https://cancergenome.nih.gov/). Patients with the survival time less than 30 days were excluded from further analyses because they may have died of other fatal complications. Ultimately, 447 patients were enrolled in our research and each tumor case corresponded to one independent patient. Data collection time: October 1, 2019.
Prognostic model construction and validation
The significance of lncRNAs for survival was analyzed using the Cox proportional hazards model by the Survival package of R (3.5.2) software. Univariate Cox analysis showed that expression of 32 lncRNAs were correlated with survival. Multivariate Cox analysis of these 32 lncRNAs revealed 8 lncRNA with significant prognostic value. We fitted all models with different variable combinations, and used Akaike Information Criterion (AIC) to evaluate goodness of fit (GOF) of each model. Finally, we selected AIC to build the model with the smallest variable. The risk score of each patient was determined according to the lncRNA expression level and the regression coefficient (β) of the weighted linear combination in the multivariate analysis. The calculation formula of risk score for each patient is as follows: Risk score =βgene1×expr(gene1)+ βgene2×expr(gene2)+...+ βgeneN×expr(geneN), (expr: lncRNA expression level), β: regression coefficient). The median risk score was used to divide the group into a low-risk and a high-risk group. To evaluate the accuracy of the prognostic model, the same algorithm was applied in the validation sets (GSE91061 and GSE98394) with survival outcomes. Melanoma patients’ mRNA sequencing data from GSE91061 and GSE98394 were downloaded using the “prefetch” software in SRA format. The SRA files were converted to fastq format data by “fastq-dump” program. “fastqc” was used to control quality. “trim_galore” was used to cut adapt. “hisat2” and “samtools” were used to generate bam files. “featureCounts” was used to get the counts of each genes. After including the clinical survival data, 44 samples from GSE98394 and 51 samples from GSE91061 were screened out for next analysis. “SVA” package in R program was used to move the batch effect in combining the two datasets.
Tumor component assessment
We use the CIBERSORT analytical tool to identify the relative percentages of 22 types of tumor infiltrating immune cells (TIICs) with normalized gene expression data [26]. The gene sets of twenty nine immune markers were defined according to the function of the immune genome [27]. The ESTIMATE (Estimation of STromal and Immune cells in MAlignant Tumors using Expression data) program was used to evaluate the immune score, stromal cell content, and tumor purity of each sample [28].
Statistical analysis
Overall survival (OS) was defined as follows: the time passed from the date of diagnosis to the end of the follow-up period, to the date of death from any cause, or to the date when patient cannot be followed-up anymore. Kaplan–Meier curves were plotted to estimate overall survival; and the log rank test was performed to evaluate statistical significance of differences in survival. Univariate and multivariate analysis was performed by Cox proportional hazards model. ROC curves and area under the curve (AUC) were calculated to determine their predictive value. Principal component analysis (PCA) was apply to observe the clustering of immune-related genes belonging to the two groups. All statistical analyses were carried out using R (3.5.2) software and P<0.05 (bilateral) was defined as a statistical difference.
Author Contributions
YW and HB performed study conception and design, acquisition of data, analysis and interpretation of data, critical revision and drafting the manuscript; XW and MZ were involved in acquisition of data, analysis and interpretation of data; CK and LT were involved in interpretation of data and critical revision; LW and HY were involved in study conception and design, interpretation of data, and drafting of manuscript.
Acknowledgments
We appreciate the TCGA database for free usage.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
Yao Wang, Li Wei, and Hua You are supported by the National Natural Science Foundation of China (81802723, 81903088, 81911530169, and 81670180). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Rigel DS. Epidemiology of melanoma. Semin Cutan Med Surg. 2010; 29:204–09. https://doi.org/10.1016/j.sder.2010.10.005 [PubMed]
- 2. Ugurel S, Röhmel J, Ascierto PA, Flaherty KT, Grob JJ, Hauschild A, Larkin J, Long GV, Lorigan P, McArthur GA, Ribas A, Robert C, Schadendorf D, Garbe C. Survival of patients with advanced metastatic melanoma: the impact of novel therapies. Eur J Cancer. 2016; 53:125–34. https://doi.org/10.1016/j.ejca.2015.09.013 [PubMed]
- 3. Fankhauser M, Broggi MA, Potin L, Bordry N, Jeanbart L, Lund AW, Da Costa E, Hauert S, Rincon-Restrepo M, Tremblay C, Cabello E, Homicsko K, Michielin O, et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci Transl Med. 2017; 9:eaal4712. https://doi.org/10.1126/scitranslmed.aal4712 [PubMed]
- 4. Radvanyi L, Pilon-Thomas S, Peng W, Sarnaik A, Mulé JJ, Weber J, Hwu P. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer—letter. Clin Cancer Res. 2013; 19:5541. https://doi.org/10.1158/1078-0432.CCR-13-1054 [PubMed]
- 5. Atianand MK, Caffrey DR, Fitzgerald KA. Immunobiology of long noncoding RNAs. Annu Rev Immunol. 2017; 35:177–98. https://doi.org/10.1146/annurev-immunol-041015-055459 [PubMed]
- 6. Chen X, Gao J, Yu Y, Zhao Z, Pan Y. Long non-coding RNA UCA1 targets miR-185-5p and regulates cell mobility by affecting epithelial-mesenchymal transition in melanoma via Wnt/β-catenin signaling pathway. Gene. 2018; 676:298–305. https://doi.org/10.1016/j.gene.2018.08.065 [PubMed]
- 7. Huang YL, Xu Q, Wang X. Long noncoding RNA DSCAM-AS1 is associated with poor clinical prognosis and contributes to melanoma development by sponging miR-136. Eur Rev Med Pharmacol Sci. 2019; 23:2888–97. https://doi.org/10.26355/eurrev_201904_17567 [PubMed]
- 8. Peng L, Chen Z, Chen Y, Wang X, Tang N. MIR155HG is a prognostic biomarker and associated with immune infiltration and immune checkpoint molecules expression in multiple cancers. Cancer Med. 2019; 8:7161–73. https://doi.org/10.1002/cam4.2583 [PubMed]
- 9. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015; 160:48–61. https://doi.org/10.1016/j.cell.2014.12.033 [PubMed]
- 10. Denaro N, Merlano MC, Lo Nigro C. Long noncoding RNAs as regulators of cancer immunity. Mol Oncol. 2019; 13:61–73. https://doi.org/10.1002/1878-0261.12413 [PubMed]
- 11. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A, Kretzer NM, Durai V, Murphy KM. Transcriptional control of dendritic cell development. Annu Rev Immunol. 2016; 34:93–119. https://doi.org/10.1146/annurev-immunol-032713-120204 [PubMed]
- 12. Hu G, Gong AY, Wang Y, Ma S, Chen X, Chen J, Su CJ, Shibata A, Strauss-Soukup JK, Drescher KM, Chen XM. LincRNA-Cox2 promotes late inflammatory gene transcription in macrophages through modulating SWI/SNF-mediated chromatin remodeling. J Immunol. 2016; 196:2799–808. https://doi.org/10.4049/jimmunol.1502146 [PubMed]
- 13. Ranzani V, Rossetti G, Panzeri I, Arrigoni A, Bonnal RJ, Curti S, Gruarin P, Provasi E, Sugliano E, Marconi M, De Francesco R, Geginat J, Bodega B, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol. 2015; 16:318–25. https://doi.org/10.1038/ni.3093 [PubMed]
- 14. Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. Elife. 2013; 2:e00762. https://doi.org/10.7554/eLife.00762 [PubMed]
- 15. Li JP, Li R, Liu X, Huo C, Liu TT, Yao J, Qu YQ. A Seven Immune-Related lncRNAs Model to Increase the Predicted Value of Lung Adenocarcinoma. Front Oncol. 2020; 10:560779. https://doi.org/10.3389/fonc.2020.560779 [PubMed]
- 16. Chen P, Gao Y, Ouyang S, Wei L, Zhou M, You H, Wang Y. A Prognostic Model Based on Immune-Related Long Non-Coding RNAs for Patients With Cervical Cancer. Front Pharmacol. 2020; 11:585255. https://doi.org/10.3389/fphar.2020.585255 [PubMed]
- 17. Profumo V, Forte B, Percio S, Rotundo F, Doldi V, Ferrari E, Fenderico N, Dugo M, Romagnoli D, Benelli M, Valdagni R, Dolfini D, Zaffaroni N, Gandellini P. LEADeR role of miR-205 host gene as long noncoding RNA in prostate basal cell differentiation. Nat Commun. 2019; 10:307. https://doi.org/10.1038/s41467-018-08153-2 [PubMed]
- 18. Cormier JN, Xing Y, Ding M, Lee JE, Mansfield PF, Gershenwald JE, Ross MI, Du XL. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006; 166:1907–14. https://doi.org/10.1001/archinte.166.17.1907 [PubMed]
- 19. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, Wong YN, Rowan A, Kanu N, Al Bakir M, Chambers T, Salgado R, Savas P, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017; 18:1009–21. https://doi.org/10.1016/S1470-2045(17)30516-8 [PubMed]
- 20. Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. 2015; 194:2985–91. https://doi.org/10.4049/jimmunol.1403134 [PubMed]
- 21. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011; 331:1565–70. https://doi.org/10.1126/science.1203486 [PubMed]
- 22. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019; 40:310–27. https://doi.org/10.1016/j.it.2019.02.003 [PubMed]
- 23. Wang W, Zhao Z, Yang F, Wang H, Wu F, Liang T, Yan X, Li J, Lan Q, Wang J, Zhao J. An immune-related lncRNA signature for patients with anaplastic gliomas. J Neurooncol. 2018; 136:263–71. https://doi.org/10.1007/s11060-017-2667-6 [PubMed]
- 24. Wei C, Liang Q, Li X, Li H, Liu Y, Huang X, Chen X, Guo Y, Li J. Bioinformatics profiling utilized a nine immune-related long noncoding RNA signature as a prognostic target for pancreatic cancer. J Cell Biochem. 2019; 120:14916–27. https://doi.org/10.1002/jcb.28754 [PubMed]
- 25. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005; 102:15545–50. https://doi.org/10.1073/pnas.0506580102 [PubMed]
- 26. Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol. 2018; 1711:243–59. https://doi.org/10.1007/978-1-4939-7493-1_12 [PubMed]
- 27. He Y, Jiang Z, Chen C, Wang X. Classification of triple-negative breast cancers based on immunogenomic profiling. J Exp Clin Cancer Res. 2018; 37:327. https://doi.org/10.1186/s13046-018-1002-1 [PubMed]
- 28. Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013; 4:2612. https://doi.org/10.1038/ncomms3612 [PubMed]