



Introduction
Elders have an increased prevalence of coronary artery disease placing them at increased risk of acute myocardial infarction [1]. Despite successful guideline-based reperfusion treatment, elders nonetheless sustain larger infarcts with greater mortality from ST elevation myocardial infarction [2, 3]. The greater susceptibility to ischemia-reperfusion injury is also observed in animal models [4, 5]. The increased cardiac susceptibility with aging in experimental models is due largely to age-induced mitochondrial dysfunction [6]. Oxidative phosphorylation (OXPHOS) is decreased in aged heart mitochondria due to impairment of the mitochondrial respiratory chain [7–10]. The dysfunctional respiratory chain increases reactive oxygen species (ROS) production [11] that sensitizes to mitochondrial permeability transition pore (MPTP) opening that in turn leads to cell death during ischemia-reperfusion [7, 12–14].
There are two populations of cardiac mitochondria that consist of subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM). SSM exist underneath the sarcolemmal membrane whereas IFM are found between myofibrils and in the perinuclear region [7, 15, 16]. Mitochondrial defects mainly occur in IFM during aging [7, 17]. Contributing mechanisms of mitochondrial dysfunction with age include oxidative modifications and deletions in mitochondrial DNA [18], oxidative modification of proteins [19], activation of mitochondrial proteases [20], the impaired removal of damaged mitochondria via mitophagy [21, 22] and an increase in endoplasmic reticulum stress [17]. Many of these mechanisms imply that aging-mediated mitochondrial dysfunction is irreversible. Previous work from our laboratory supported the intriguing finding that the defects in aged heart mitochondria can be reversed [6, 17]. Furthermore, treatment of animals during the progression of aging can lead to the attenuation of age-related cardiac mitochondrial dysfunction [8]. Thus, the aged heart is not condemned to sustain greater injury due to the presence of dysfunctional mitochondria. Treatment of aged rats with the small molecule metabolite acetylcarnitine in the baseline condition improved mitochondrial function [6], supporting that the aging-induced mitochondrial defect is potentially reversible. Following an improvement in the previously established age-related mitochondrial dysfunction, aged rat hearts sustained substantially less injury during a subsequent ischemia-reperfusion stress [6]. These results indicate that reversing mitochondrial defects in aged hearts is possible and could be a promising strategy to attenuate cardiac injury during ischemia and reperfusion [6, 23].
Mitochondria are in close contact with the endoplasmic reticulum (ER) [24, 25]. ER dysfunction (ER stress) is one of the factors that induces mitochondrial defects in adult hearts [26, 27]. Induction of acute ER stress using thapsigargin (a calcium-ATPase inhibitor) impairs mitochondrial function in adult hearts as shown by decreased oxidative phosphorylation and decreased respiratory enzyme activities [26, 28], and a greater sensitization to MPTP opening [17, 26, 27, 29]. Interestingly, aging leads to increased ER stress that occurs earlier than the onset of mitochondrial dysfunction during aging [17]. These results suggest that the ER stress leads to mitochondrial dysfunction in aged hearts [17].
Metformin, an anti-diabetic drug, reduces heart injury during ischemia-reperfusion by activating AMP-activated protein kinase (AMPK) signaling [26, 30, 31]. The activation of AMPK reduces cell injury during oxidative stress via decreased MPTP opening [26, 32]. Treatment with metformin in higher dose decreases cardiac injury through inhibition of complex I during early reperfusion [33]. Metformin treatment also decreases ER stress. Angiotensin II-induced ER stress is decreased with metformin treatment by activating AMPK [26, 34]. Metformin decreases β-cell lipotoxicity by a decrease in ER stress [35]. Metformin treatment improved mitochondrial function following thapsigargin-induced acute ER stress [26]. Thus, we tested the hypothesis that preexisting ER stress in the aged hearts can be attenuated with chronic metformin treatment. If ER stress was indeed decreased, then the contribution of a reduction in ER stress to an improvement in the preexisting age-induced cardiac mitochondrial dysfunction was evaluated. Finally, the relationship of the reversal of mitochondrial defects in aged hearts to the extent of cardiac injury during subsequent ischemia and reperfusion in the high-risk aged heart was challenged.
Results
Metformin increased the phosphorylation of AMPK in aged hearts
Metformin treatment increases the phosphorylation of AMPK (Thr172) in adult mice compared to vehicle [26]. Compared to vehicle treatment, metformin treatment increased the phosphorylation of AMPK and acetyl-CoA carboxylase (Ser70) (ACC) in 24 mo. hearts (Figure 1B). ACC is phosphorylated by activated AMPK and is an index of functional AMPK activation [31]. Thus, metformin treatment also activates AMPK in the aged hearts [26].
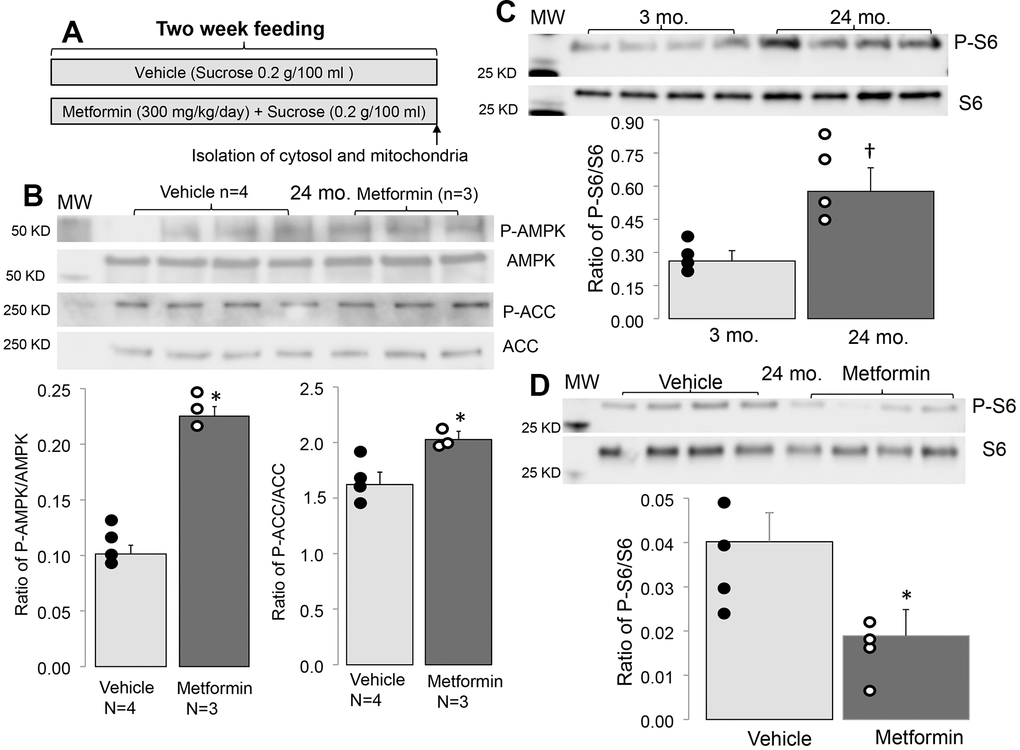
Figure 1. Administration of metformin increased phosphorylation of AMPK and ACC in aged mouse hearts. (A) Shows the protocol of metformin feeding. In metformin treated groups, metformin (300 mg/kg/day body weight) was dissolved in drinking water with sucrose (0.2g/100 ml) as sweetener and fed to mice for 2 weeks. In vehicle treated groups, mice were fed with drinking water with added sucrose (0.2g/100 ml). Compared to vehicle, metformin treatment increased the phosphorylation of AMPK and ACC in aged hearts, supporting that metformin feeding activates the AMPK in the aged hearts (B). The phosphorylation of protein S6 was increased with age, indicating an increased activity of mTORC1 (C). Metformin treatment decreased the age induced S6 phosphorylation (D). Mean ± SEM. *p<0.05 vs. vehicle, †p<0.05 vs. 3 mo.
Metformin decreased mTOR activation in 24 mo. mice
The AMPK activation by metformin decreases the activity of mTOR (mechanistic target of rapamycin activity) [26, 36]. mTOR includes two complexes: one is mTORC1 (mTOR complex 1), and the second is mTORC2 (mTOR complex 2) [37]. Since mTORC1 is linked to ER stress by the unfolded protein response [38], the mTORC1 activation state was assessed using the phosphorylation state of ribosomal protein S6 in 24 mo. mice [17]. Compared to 3 mo., phosphorylated S6 content was significantly increased in 24 mo. hearts (Figure 1C). Metformin treatment decreased the phosphorylation of S6 in 24 mo. mice compared to vehicle (Figure 1D), suggesting that metformin treatment leads to mTORC1 inhibition in 24 mo. mice.
Metformin decreased ER stress in the aged hearts
The induction of ER stress increased CCAAT/enhancer-binding protein homologous protein (CHOP) content and the cleavage of ATF6 (cleaved activated transcription factor 6) [17, 39]. Therefore, CHOP and cleaved ATF6 are a robust indicator of ER stress [17, 39]. The contents of the cleaved ATF6 and CHOP were elevated in 24 mo. versus 3 mo. mice (Figure 2A, 2B), indicating an increased ER stress in aged hearts. Metformin treatment decreased the contents of cleaved ATF6 and CHOP in 24 mo. hearts (Figure 2A, 2B), supporting that metformin treatment decreases the ER stress that occurs during aging.
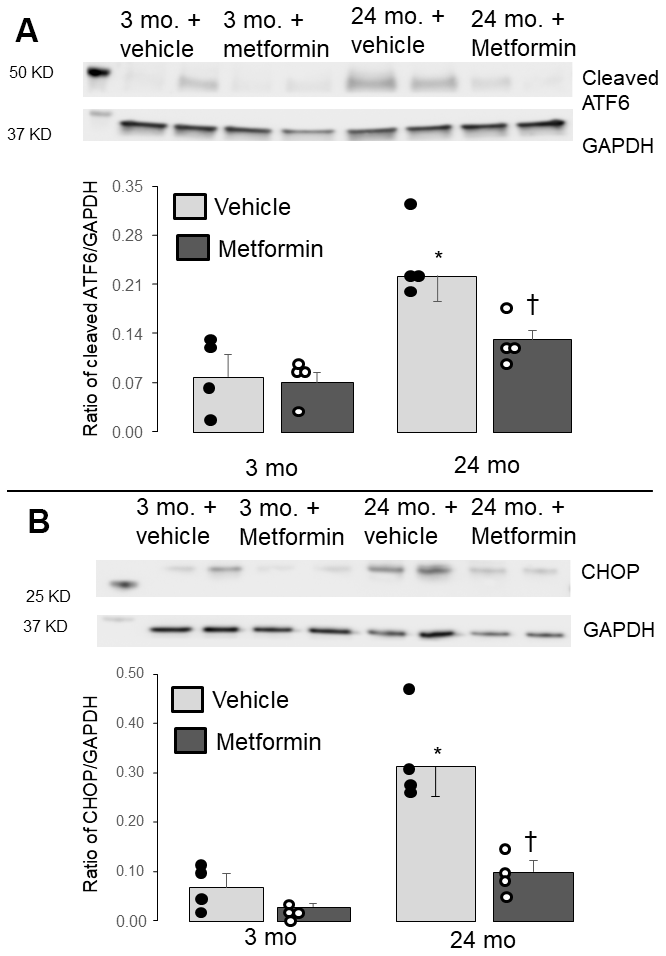
Figure 2. Administration of metformin decreased the endoplasmic reticulum (ER) stress in aged mouse hearts. Compared to 3 mo., the contents of the cleaved ATF6 (A) and CHOP (B) were significantly increased in 24 mo., supporting the presence of increased ER stress in aged hearts. The contents of cleaved ATF6 and CHOP were markedly decreased in metformin-treated 24 mo. hearts compared to vehicle, supporting that metformin treatment decreased the ER stress present in aged hearts. Metformin treatment did not alter the ER stress in 3 mo. hearts. Mean ± SEM. *p<0.05 vs. 3 mo. vehicle, †p<0.05 vs. 24 mo. vehicle. n=4 in each group.
Metformin improved oxidative phosphorylation in aged IFM
Aging leads to electron transport chain defects in IFM [40]. Compared to 3 mo., the rate of state 3 respiration in IFM was decreased in 24 mo. in the presence of complex I substrates (glutamate + malate) (Table 1) [17]. The maximal rate of ADP-stimulated respiration was also decreased in IFM from 24 mo. (Table 1). The ADP-limited respiration (state 4) in IFM was also slightly decreased in 24 mo. (Table 1). The uncoupled respiration stimulated with dinitrophenol was decreased in 24 mo. IFM (Figure 1C), localizing the defect to the electron transport chain [41].
Table 1. The rate of oxidative phosphorylation in IFM from young and aged mice with or without metformin treatment.
| Mice | 3 mo. | 24 mo. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Groups | Vehicle (n=13) | Metformin (n=5) | Vehicle (n=11) | Metformin (n=5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex I substrates: glutamate + malate | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| State 3 (nAO/min/mg) | 356±13 | 367±14 | 271±13* | 337±22† | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| State 4 (nAO/min/mg) | 58±3 | 58±5 | 48±1* | 58±3† | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RCR | 6.3±0.2 | 6.6±0.5 | 5.7±0.2 | 5.9±0.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 mM ADP (nAO/min/mg) | 481±17 | 487±29 | 318±19* | 397±32† | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex II substrates: Succinate + rotenone | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| State 3 (nAO/min/mg) | 860±30 | 735±13* | 779±34 | 847±30 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| State 4 (nAO/min/mg) | 256±12 | 226±11 | 212±8* | 218±8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RCR | 3.4±0.1 | 3.3±0.1 | 3.7±0.1 | 3.9±0.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 mM ADP (nAO/min/mg) | 849±29 | 746±19* | 769±34 | 788±32 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mean ± SEM. *p<0.05 vs. 3 mo. Vehicle, †p<0.05 vs. 24 mo. Vehicle. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The state 3 respiration in IFM was not decreased in 24 mo. with succinate as complex II substrate using rotenone to block potential reverse electron flow (Table 1) [42, 43]. The rate of state 4 respiration in IFM was decreased in 24 mo. using succinate (Table 1). The high ADP-stimulated respiration was not decreased in 24 mo. IFM compared to 3 mo. with complex II substrates (Table 1), suggesting that the maximal rate of respiration with complex II substrate was not altered in aged IFM. These results localize the electron transport defect with aging predominantly to complex I in murine IFM [17].
Metformin treatment improved state 3 respiration in 24 mo. IFM in the presence of either complex I or II substrates (Table 1). Metformin treatment also improved the dinitrophenol-uncoupled respiration in 24 mo. IFM with complex I substrate (Figure 3B), supporting an improvement in the age-induced defect in electron transport. Metformin also increased state 4 respiration in 24 mo. IFM (Table 1). Importantly, MET did not alter mitochondrial respiration in 3 mo. mice without a defect in OXPHOS.
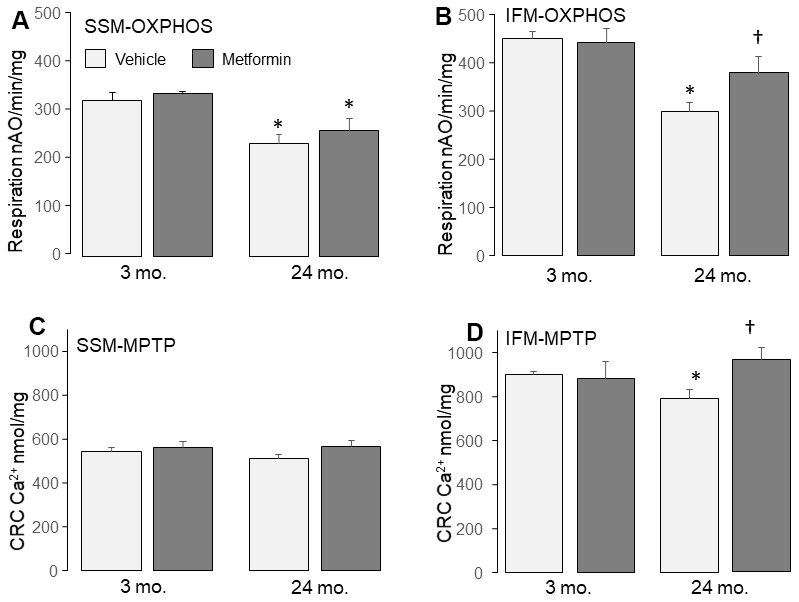
Figure 3. Administration of metformin improved mitochondrial function in aged IFM. Compared to 3 mo., dinitrophenol (DNP) uncoupled respiration was decreased in 24 mo. SSM (A) and IFM (B) using complex I substrate, supporting that aging impairs the mitochondrial respiratory chain. Metformin feeding improved oxidative phosphorylation in 24 mo. IFM oxidizing complex I substrates (B). Metformin feeding did not affect the oxidative phosphorylation in 24 mo. SSM with complex I substrates (A). Compared to 3 mo., the calcium retention capacity (CRC) was decreased in 24 mo. IFM (D), supporting that aging sensitizes to mitochondrial permeability transition pore (MPTP) opening. Metformin feeding improved the CRC in 24 mo. IFM (D) but not in 24 mo. SSM (C), indicating that metformin feeding decreased MPTP opening in 24 mo. IFM. Mean ± SEM; * p <0.05 vs. 3 mo. vehicle. †p<0.05 vs. 24 mo. vehicle. n=13 in 3 mo. vehicle group. N=5 in 3 mo. metformin treatment group. n=10 in 24 mo. vehicle group. N=9 in 24 mo. metformin treatment group.
Metformin decreased the age-induced MPTP opening in IFM
A decrease in calcium retention capacity (CRC) is an indicator of an increased sensitivity to MPTP opening in mitochondria [26, 44]. The CRC was significantly decreased in 24 mo. IFM compared to 3 mo. (Figure 3D), supporting that aging sensitizes to MPTP opening in IFM. Metformin led to a decreased sensitivity to MPTP opening in IFM from 24 mo. hearts (Figure 3D). These results indicate that metformin treatment lessens the probability of MPTP opening in aged IFM.
Metformin decreased cardiac injury following ischemia-reperfusion
Metformin feeding did not affect heart or body weight in 24 mo. old mice (Table 3). Metformin treatment did not alter left ventricular developed pressure (LVDP) [47] (Table 3) or end diastolic pressure (LVEDP) (Table 3) before ischemia in 24 mo. hearts compared to vehicle. Ischemia followed by reperfusion led to decreased LVDP and increased LVEDP in both vehicle and metformin-treated hearts compared to the pre-ischemia value (Table 3). However, metformin treatment markedly decreased the infarct size compared to vehicle (Figure 4B), supporting that metformin treatment that leads to the restoration of mitochondrial function mitigates cardiac injury in aged hearts during subsequent in vitro ischemia and reperfusion.
Table 3. Hemodynamic change during ischemia-reperfusion in 24 mo. hearts with or without metformin treatment.
| Vehicle (n=9) | Metformin (n=10) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Body weight (g) | 33.1 ± 1.5 | 33.6 ± 1.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heart weight (g) | 0.17 ± 0.01 | 0.18 ± 0.01 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ratio of Heart/body | 0.0050 ± 0.0003 | 0.0053 ± 0.002 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pre-Ischemia | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LVDP (mmHg) | 82 ± 8 | 73 ± 2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LVEDP (mmHg) | 5 ± 1 | 6 ± 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| End of Reperfusion | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LVDP (mmHg) | 39 ± 5* | 30 ± 7* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LVEDP (mmHg) | 32 ± 6* | 34 ± 7* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mean ± SEM. *p<0.05 vs. corresponding pre-ischemic value. All p=NS vehicle vs. metformin. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
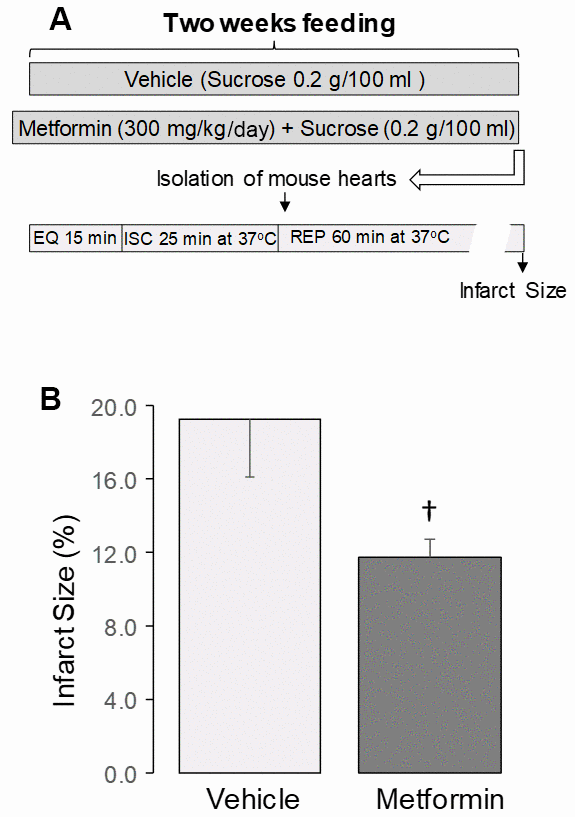
Figure 4. Administration of metformin decreased cardiac injury in aged mouse hearts. (A) Shows the experimental protocol. Metformin feeding was the same as in Figure 1. Isolated perfused hearts received no ex vivo treatment. The isolated mouse hearts underwent 25 min. global ischemia at 37° C and 60 min. reperfusion. Metformin treatment decreased the infarct size in aged 24 mo. hearts compared to vehicle (B), supporting that metformin treatment decreased cardiac injury in 24 mo. hearts following ischemia-reperfusion. Mean ± SEM. †p<0.05 vs. 24 mo. vehicle. n=9 in vehicle treated group. n=10 in metformin treated group.
Discussion
In the present study, we show that chronic treatment for two weeks with metformin improved pre-existing age-induced ER stress (Figure 2) and mitochondrial dysfunction (Figure 3B). Next, we asked if the relief of ER stress with improved mitochondrial function led to decreased cardiac injury following ischemia and reperfusion. In the buffer perfused aged heart, infarct size was substantially reduced (Figure 4D). As expected, metformin feeding increased AMPK activation as shown by the phosphorylation of both AMPK and ACC (Figure 1). Thus, a translational relevant approach can attenuate metabolic defects present in the aged heart and reduce injury in the aged heart from superimposed cardiac stress [41]. Furthermore, this treatment approach uses the novel approach to decrease cardiac injury from ischemia and reperfusion by the treatment of preexisting age-induced disease.
OXPHOS is mainly impaired in IFM from aged hearts [7]. A decrease in dinitrophenol-uncoupled respiration supports that the defects in aged heart mitochondria are located in the electron transport chain [7, 40, 48]. Aging also sensitizes to MPTP opening mainly in IFM [14]. The results support previous findings that the aging defect predominantly involves IFM. The ER stress increases in aged hearts [17, 49]. Metformin attenuates the ER stress in aged hearts indicated by decreased CHOP and cleaved ATF6 contents (Figure 2). CHOP is a major endpoint of the activation of ER stress, with increased formation not only via cleavage of ATF6, but also via phosphorylation and activation of PERK (RNA-activated protein kinase-like ER kinase) [50] as well as the activation of IRE1α (inositol requiring enzyme 1) [51–55]. Concomitant with the reduction in ER stress, metformin treatment improves OXPHOS and decreases the sensitivity of MPTP opening in aged IFM (Figure 3D). These results support that the increased ER stress causes mitochondrial dysfunction in the aged heart [17]. Restoration of mitochondrial function prior to the onset of ischemia and reperfusion in aged hearts by metformin, in turn, decreases cardiac injury during ischemia-reperfusion. Thus, chronic metformin treatment could be a promising strategy to protect aged hearts during ischemia and reperfusion by reducing the ER stress present in the baseline state.
Chronic metformin pretreatment decreases infarct size in adult mice [56]. Furthermore, ongoing metformin treatment appears beneficial in diabetic patients who suffer a subsequent infarct [57]. Metformin treatment in diabetic patients substantially improves cardiovascular disease outcomes in the UK Prospective Diabetes Study (UKPDS) [58]. Metformin protects in experimental models of heart failure [57]. Metformin is well tolerated in non-diabetic mice [33, 59]. The GIPS-III trial found that metformin therapy was also safe in non-diabetic adult patients, including those suffering an acute myocardial infarction [60]. Thus, there is a substantial potential for a repurposing in elders to attenuate age-induced susceptibility of the heart.
The mechanisms by which aging leads to mitochondrial dysfunction remain unclear. In the present study, we find that ER stress is markedly elevated in aged hearts. In addition, the reduction of ER stress using metformin improves mitochondrial function in aged hearts. The results support that the increased ER stress causes mitochondrial dysfunction during aging. Metformin treatment decreased ER stress in other disease models including catecholamine stress [61], pressure overload [34] and diabetes [62]. However, the mechanisms of action of metformin from these studies also remains to be better defined. Metformin may decrease ER stress due to modulation of SERCA activity in the adult heart [26, 63]. To our knowledge, the improvement in mitochondrial function in the aged heart with attenuation of the ER stress by metformin therapy has not been previously described.
Two weeks of metformin therapy markedly reduced ER stress and subsequent mitochondrial dysfunction in the adult heart [26, 28]. This previous work from our laboratory [26] demonstrates that metformin decreases the thapsigargin-induced ER stress in adult hearts with the protection of mitochondrial oxidative phosphorylation and complex I of the respiratory chain [26, 28]. In the present study, metformin can decrease the age-induced pre-existing ER stress present in aged hearts before the occurrence of other cardiac disease. These results support that metformin is an option to decrease the ER stress in aged hearts before the onset of a superimposed stress such as ischemia and reperfusion. Administration of the AMPK activator 5-aminoimidazole-4-carboxyamide-1-beta-D-ribofuranoside decreases the ER stress in cardiac myocytes during hypoxia-reoxygenation [64], indicating that activation of AMPK decreases the ER stress. Metformin is clearly a more translational relevant approach to modulate ER stress and improve mitochondrial function than the classic small molecule chaperone 4-phenylbutyrate (4-PBA) that is commonly used to inhibit ER stress in animal studies [65], including during aging [17, 66, 67]. To further support the role of age-induced ER stress in mitochondrial dysfunction during aging, previous work showed that two weeks of treatment with the 4-PBA reduced ER stress and improved pre-existing age-induced mitochondrial dysfunction [17, 68].
Metformin is a traditional anti-diabetic drug with cardioprotective effects by activating AMPK signaling [69]. The mechanism of AMPK activation is due to subtle complex I inhibition, leading to a modest decrease in energy charge. This inhibition is observed with chronic metformin therapy and requires only micromolar intracellular concentration [70, 71]. Metformin, via this AMPK activation, leads to enhanced mitophagy and mitochondrial biogenesis [72–74]. Metformin downregulates the mechanistic target of rapamycin through activation of AMPK [36, 75]. In addition to the relief of ER stress, the potential mechanisms of AMPK protection include favoring glucose uptake and oxidation, modulation of autophagy [76], and augmentation of mitochondrial biogenesis [77]. In the present study, chronic metformin treatment activates AMPK in aged hearts as shown by increased phosphorylation of the AMPK and ACC. These results suggest that metformin decreases the ER stress during aging by activating the AMPK signaling. Interestingly, a recent study shows that the decreased PGC1-α level in aged hearts is not dependent on AMPK activity. Metformin treatment does improve mitochondrial biogenesis in aged hearts [78]. These results suggest that metformin-mediated improvement in biogenesis is not solely dependent on AMPK activation.
Activation of AMPK by metformin leads to decreased mTOR activity through activation of AMPK [36, 37]. The mTORC1 performs the classic functions of mTOR including nutrient sensing, regulation of protein synthesis, and autophagy [37], whereas the mTORC2 is involved in cell proliferation and insulin signaling [37]. mTORC1 is linked to ER stress via the unfolded protein response [38]. Inhibition of mTOR extends lifespan [79]. mTORC1 regulates the translation of several mitochondria-related mRNAs including components of complex I and V and transcription factor A (TFAM) [80]. Inhibition of mTORC1 in 24 mo. mice improves cardiovascular function and reverses cardiac fibrosis [81] by improving mitochondrial function [82]. The inhibition of mTORC1 by activation of AMPK itself prolongs lifespan [82, 83]. The current study shows that metformin treatment results in activation of AMPK in 24 mo. old mice accompanied by decreased mTORC1 activation indicated by decreased phosphorylation of the protein S6. These results support the notion that activation of AMPK provides protective benefits in aged hearts by inhibiting mTORC1.
AMPK activation is enhanced by the sestrin protein family [84]. Sestrin2 is a stress-induced scaffold protein that mediates AMPK activation via interaction with LKB1 [5, 85, 86]. Sesn2 expression at baseline was reduced in aged hearts [5, 86] and a decrease in Sesn2 attenuates AMPK activation. Sesn2 mediated AMPK activation also leads to the downregulation of mTOR [87, 88] that can protect cells against ER stress by decreasing protein synthesis [89, 90]. The age-induced decrease in Sesn2 expression may impair activation of AMPK in response to stress. However, in the current study, chronic metformin therapy was nonetheless able to activate AMPK to a sufficient extent to reduce ER stress and improve mitochondrial function, similar to protection against ER stress in the adult heart [26].
In addition to increased cardiac injury during ischemia-reperfusion, aging also invalidates endogenous signaling of cardiac protection that leads to many cardioprotective approaches that protect younger adult hearts, leading these approaches to fail in aged hearts [7, 12, 91]. Treatment of aged rats with the small molecule metabolite acetylcarnitine improved mitochondrial function [6], supporting the potential reversibility of the mitochondrial defect in aging. This concept is further supported by current findings that metformin restores mitochondrial function in the aged hearts. Treatment with acetylcarnitine in the aged heart not only restored mitochondrial function, but also reduced cardiac injury during ischemia-reperfusion [6], indicating that the age-induced mitochondrial defects contribute to cardiac injury [6]. The current study shows that metformin treatment improves mitochondrial function and decreases cardiac injury in aged hearts during ischemia and reperfusion, expanding available options to restore mitochondrial function in aged hearts. Metformin treatment may provide additional benefits in the high-risk elderly population with a greater incidence of myocardial infarction [2, 3] that suffer substantially greater cardiac injury and decreased survival should an infarction occur [2, 3].
MPTP opening leads to cell death during ischemia and reperfusion [92]. Increased ER stress favors MPTP opening in adult heart mitochondria [26–28]. In the present study, aging leads to increased sensitivity to MPTP opening mainly in IFM. Importantly, attenuation of the ER stress with metformin decreases MPTP opening in mitochondria isolated from aged hearts. The current study provides direct evidence that the increased ER stress favors MPTP opening during aging. ER stress mediated oxidative [93] and calcium-driven [27] mechanisms impact mitochondria and likely contribute to the increased susceptibility to MPTP opening even in the baseline state. Metformin treatment decreases the MPTP opening during acute ER stress by activating AMPK [26]. This mechanism may be also involved in decreased MPTP opening in aged hearts with metformin treatment.
Limitations
The current study emphasized a translational relevant approach to activate AMPK driven reduction in ER stress in the baseline aging condition to improve mitochondrial function. The response from aged hearts paralleled the response of metformin therapy to reduce ER stress, likely via AMPK mediated mechanisms. The period of pretreatment that improved mitochondrial function in the current study most certainly resulted in modest inhibition of complex I that was present during ischemia and reperfusion, even in the absence of additional metformin treatment [70, 71] which would have exerted protection in aged heart during ischemia [94] and early reperfusion [11]. Although inhibition of complex I by metformin leads to deceased cardiac injury during ischemia-reperfusion [33], other mechanisms may be also involved in the protection of metformin. The improvement of OXPHOS, especially with complex I substrates, does not suggest that substantial complex I inhibition was present before ischemia and reperfusion from the chronic metformin treatment, though a beneficial impact regarding production of reactive oxygen species production and attenuation of permeability transition pore opening cannot be excluded [70]. Metformin may decrease cardiac injury by decreasing sensitivity to MPTP opening as shown in cultured cells and isolated mitochondria [71]. ROS generated by reverse electron flow contributes to cardiac injury during reperfusion [95]. Metformin treatment likely also mitigates cardiac injury through reduction of the reverse flow-induced ROS generation [70]. Activation was sufficient despite age-related decrements in sestrin 2 content, the role of which will also require further study. The mechanisms of the ER and mitochondrial interactions that favor age-induced mitochondrial dysfunction will require additional work. Finally, the model of ischemia and reperfusion is one of acute injury, the study of longer-term reperfusion and recovery periods, especially the in vivo ischemia-reperfusion model, will be required in the future.
Conclusions
Chronic treatment of aged mice for two weeks with metformin activated AMPK signaling leading to a reduction in age-induced ER stress with substantial improvement of function in heart mitochondria (Figure 5). In the setting of these improvements, the age-enhanced susceptibility to cardiac injury during myocardial infarction and reperfusion was improved. The current study advances the understanding of mechanisms involved in aging-mediated mitochondrial dysfunction, but also provides a novel treatment opportunity to restore mitochondrial function in aging in that the ER stress can be modulated by pharmacologic interventions. This translational treatment approach provides key evidence for a new treatment paradigm to decrease injury from superimposed cardiac disease in the high-risk aged heart by the treatment of age-related defects to restore mitochondrial function before the onset of acute cardiac disease in the elderly heart. This approach is potentially relevant to acute coronary syndromes with ST elevation-induced myocardial infarction with an increased risk of progression to heart failure [2, 96], acute coronary syndromes with non-ST elevation infarction [97, 98], heart failure with preserved ejection fraction [99, 100], and chemotherapy cardiotoxicity [101].
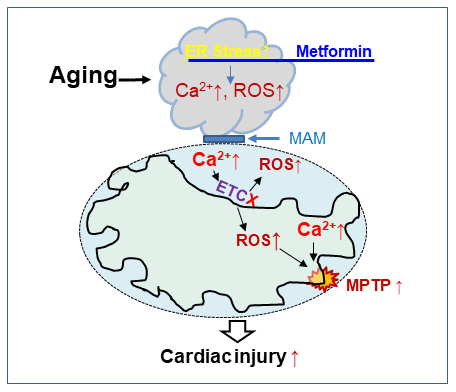
Figure 5. The chronic administration of metformin decreases endoplasmic reticulum stress with improvement in cardiac mitochondrial function in aged mouse hearts. Aging increases endoplasmic reticulum (ER) stress that causes mitochondrial dysfunction by increasing calcium overload and ROS generation. The ER and cardiac mitochondrial interact via mitochondrial associated membranes (MAM). An increase in mitochondrial calcium overload and ROS generation sensitizes to mitochondrial permeability transition pore (MPTP) opening that augments cardiac injury during ischemia-reperfusion. Metformin treatment decreases cardiac injury by restoring mitochondrial function before ischemia through attenuation of the ER stress in the aged hearts.
Materials and Methods
Metformin treatment
The Animal Care and Use Committees of Virginia Commonwealth University and the McGuire Department of Veterans Affairs Medical Center approved the study. Male mice of young adult (3 mo.) and aged mice (24 mo.) were used in this study. Mice were given a normal diet with ad libitum access to food and water throughout the experiment. Normal diet included 16% protein and 4% fat. In metformin treated mice, metformin (300 mg/kg/day body weight) was dissolved in drinking water with sucrose (0.2g/100 ml) as sweetener and fed to mice for 2 weeks [56] (Figure 1A). The dose of metformin was based upon previous studies in the rat [56]. Control mice received drinking water with sucrose vehicle (0.2g/100 ml). Deep anesthesia was induced in mice with pentobarbital sodium (100 mg/kg, i.p.). Then, mitochondria were isolated from the excised mouse heart.
Isolation of cytosol, mitochondria, and nucleus
SSM and IFM were isolated as previously described [17]. The mouse heart was first placed in cold buffer A (composition in mM: 100 KCl, 50 MOPS [3-(N-morpholino) propanesulfonic acid], 1 EGTA, 5 MgSO4, and 1 ATP). The heart was blotted dry, weighed, and homogenized using a polytron tissue homogenizer at 10,000 rpm for 2.5 seconds. The polytron homogenate was first centrifuged at 500 g for 10 min. The supernatant was used to isolate SSM with further centrifugation at 3,000 g for 10 min. The pellet from the 500 g centrifuge step was washed and used for IFM isolation. The skinned myofibers, obtained from the polytron homogenization step, were resuspended in buffer A and incubated with 5 mg/g (wet weight) trypsin for 10 min at 4° C. Then, buffer B [buffer A including 0.2% bovine serum albumin (BSA)] was added to stop trypsin effect. The crude SSM and IFM were washed with buffer B. The purified SSM and IFM were suspended in 80 mM KCl, 50 mM MOPS, and 0.5 mM EGTA for functional measurement.
Oxygen consumption in isolated mitochondria was measured using a Clark-type oxygen electrode at 30° C as previously described [102]. Glutamate (20 mM) + Malate (10 mM) were used as complex I substrate. Succinate (20 mM) was used as the complex II substrate with the inclusion of 7.5 μM rotenone. ADP (2 mM) was used to determine the maximal rate of ADP-stimulated respiration.
Calcium retention capacity (CRC) in isolated mitochondria
The CRC was used to assess the calcium induced MPTP opening in freshly isolated SSM and IFM [103]. The assay medium included mitochondria (125 μg/ml), 150 mM sucrose, 50 mM KCl, 2 mM KH2PO4, and 5 mM succinate in 20 mM Tris/HCl with pH at 7.4. Calcium Green-5N (0.5 uM, Thermo Scientific, Waltham, MA) was used to monitor extra-mitochondrial Ca2+ concentration with excitation and emission wavelengths set at 500 and 530 nm, respectively [103]. Sequential exogenous calcium (5 nmol/pulse) was added into cuvettes until MPTP opening occurred, shown by a burst release of calcium from mitochondria.
Measurement of ROS in SSM and IFM
The amount of H2O2 generation in SSM and IFM was measured using Amplex red as a fluorogenic indicator in the presence of horseradish peroxidase. Freshly isolated SSM or IFM (200 μg) were incubated in chelex-treated buffer [pH 7.4 (150 mM KCl, 5 mM KH2PO4, 1 mM EGTA)] in the presence of 25 μM Amplex Red and 0.20 units/ml HRP. Glutamate + malate was used as complex I substrate, and succinate + rotenone was used as complex II substrate. Rotenone (complex I inhibitor) and antimycin A (complex III inhibitor) were used to induce maximal H2O2 generation from complex I and complex III, respectively [104].
Isolated perfused heart model of Ischemia and Reperfusion
Mouse hearts were excised under deep anesthesia using pentobarbital sodium (100 mg/kg i.p.) and anticoagulated with heparin (1,000 IU/kg i.p.). The isolated heart is mounted in the Langendorff setup and perfused with modified Krebs-Henseleit (K-H) buffer oxygenated with 95% O2-5% CO2 through aorta. A balloon was inserted into the left ventricle to monitor cardiac function. The heart was first perfused with K-H buffer for 15 min. The hearts underwent 25 min. of global ischemia at 37° C and 60 min. of reperfusion (Figure 4A). In order to keep a constant heart rate, hearts were paced at 420 beats/min during the 15 min. equilibration period and after 10 min. of reperfusion [105]. Myocardial infarct size was measured at the end of the reperfusion using staining with triphenyl tetrazolium chloride (TTC) [106].
Western blotting
Proteins from mitochondria or cytosol were separated using 12% or 4-15% Tris-glycine gels (Bio-Rad, Hercules, CA) and transferred to a PVDF membrane (Millipore) using semi-dry transfer (Bio-Rad). The membrane was incubated for 1 hour at room temperature in 5% (w/v) non-fat dry milk (Bio-Rad) in TBS-T buffer (10 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween-20). Then, the membrane was incubated with primary antibody overnight at 4° C. After 1 hour incubation at room temperature with a 1:10,000 dilution of HRP-conjugated anti-mouse or anti-rabbit IgG F(ab)2 (GE Healthcare Life Sciences, Piscataway, NJ), blots were developed using ECL Plus Western Blotting Detection Reagents (GE Healthcare Life Sciences, Piscataway, NJ) [28, 47].
Statistical analysis
Data are expressed as the mean ± standard error [107]. Normality distribution was assessed. One-way analysis of variance (ANOVA) was used to compare the differences between groups (≥ 3 groups). The Student-Newman-Keuls test of multiple comparisons was used when a significant F value was obtained. The non-paired student test (two tails) was to compare the differences between two groups. Statistical significance was accepted when a value of p less than 0.05 was obtained.
Author Contributions
QC-designed experiments, performed experiments, analyzed data, wrote the manuscript, obtained research funding; JT-performed experiments, analyzed data; YH-performed experiments, analyzed data; EJL-designed experiments, analyzed data, wrote the manuscript, obtained research funding.
Acknowledgments
The authors have read the journal’s authorship agreement and the manuscript has been reviewed and approved by all named authors.
Conflicts of Interest
All authors have read the journal’s policy on authorship agreement and disclosure of potential conflicts of interest and have none to declare.
Funding
This work was supported by the Office of Research and Development, Medical Research Service Merit Review Award (2IO1BX001355-01A2), Department of Veterans Affairs (QC, JT, YH, EJL), the National Institutes of Aging (1R21AG054975-01) (QC, JT), VCU’s CTSA (UL1TR000058 from the National Institutes of Health's National Center for Advancing Translational Science) (JT, QC, EJL), Pauley Heart Center Grant (QC) and the Pauley Heart Center, Virginia Commonwealth University (QC, EJL).
References
- 1. Owlia M, Dodson JA, King JB, Derington CG, Herrick JS, Sedlis SP, Crook J, DuVall SL, LaFleur J, Nelson R, Patterson OV, Shah RU, Bress AP. Angina severity, mortality, and healthcare utilization among veterans with stable angina. J Am Heart Assoc. 2019; 8:e012811. https://doi.org/10.1161/JAHA.119.012811 [PubMed]
- 2. Lesnefsky EJ, Lundergan CF, Hodgson JM, Nair R, Reiner JS, Greenhouse SW, Califf RM, Ross AM. Increased left ventricular dysfunction in elderly patients despite successful thrombolysis: the GUSTO-I angiographic experience. J Am Coll Cardiol. 1996; 28:331–37. https://doi.org/10.1016/0735-1097(96)00148-9 [PubMed]
- 3. Lattuca B, Kerneis M, Zeitouni M, Cayla G, Guedeney P, Collet JP, Montalescot G, Silvain J. Elderly patients with ST-segment elevation myocardial infarction: A patient-centered approach. Drugs Aging. 2019; 36:531–39. https://doi.org/10.1007/s40266-019-00663-y [PubMed]
- 4. Lesnefsky EJ, Gallo DS, Ye J, Whittingham TS, Lust WD. Aging increases ischemia-reperfusion injury in the isolated, buffer-perfused heart. J Lab Clin Med. 1994; 124:843–51. [PubMed]
- 5. Quan N, Wang L, Chen X, Luckett C, Cates C, Rousselle T, Zheng Y, Li J. Sestrin2 prevents age-related intolerance to post myocardial infarction via AMPK/PGC-1α pathway. J Mol Cell Cardiol. 2018; 115:170–78. https://doi.org/10.1016/j.yjmcc.2018.01.005 [PubMed]
- 6. Lesnefsky EJ, He D, Moghaddas S, Hoppel CL. Reversal of mitochondrial defects before ischemia protects the aged heart. FASEB J. 2006; 20:1543–45. https://doi.org/10.1096/fj.05-4535fje [PubMed]
- 7. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. 2016; 118:1593–611. https://doi.org/10.1161/CIRCRESAHA.116.307505 [PubMed]
- 8. Quarles E, Basisty N, Chiao YA, Merrihew G, Gu H, Sweetwyne MT, Fredrickson J, Nguyen NH, Razumova M, Kooiker K, Moussavi-Harami F, Regnier M, Quarles C, et al. Rapamycin persistently improves cardiac function in aged, male and female mice, even following cessation of treatment. Aging Cell. 2020; 19:e13086. https://doi.org/10.1111/acel.13086 [PubMed]
- 9. Moghaddas S, Hoppel CL, Lesnefsky EJ. Aging defect at the QO site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 2003; 414:59–66. https://doi.org/10.1016/s0003-9861(03)00166-8 [PubMed]
- 10. Dai DF, Liu Y, Basisty N, Karunadharma P, Dastidar SG, Chiao YA, Chen T, Beyer RP, Chin MT, Maccoss M, La Spada AR, Rabinovitch PS. Differential effects of various genetic mouse models of the mechanistic target of rapamycin complex I inhibition on heart failure. Geroscience. 2019; 41:847–60. https://doi.org/10.1007/s11357-019-00119-6 [PubMed]
- 11. Chen Q, Ross T, Hu Y, Lesnefsky EJ. Blockade of electron transport at the onset of reperfusion decreases cardiac injury in aged hearts by protecting the inner mitochondrial membrane. J Aging Res. 2012; 2012:753949. https://doi.org/10.1155/2012/753949 [PubMed]
- 12. Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL. Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annu Rev Pharmacol Toxicol. 2017; 57:535–65. https://doi.org/10.1146/annurev-pharmtox-010715-103335 [PubMed]
- 13. Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC
Jr , Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013; 18:239–50. https://doi.org/10.1016/j.cmet.2013.07.002 [PubMed] - 14. Hofer T, Servais S, Seo AY, Marzetti E, Hiona A, Upadhyay SJ, Wohlgemuth SE, Leeuwenburgh C. Bioenergetics and permeability transition pore opening in heart subsarcolemmal and interfibrillar mitochondria: effects of aging and lifelong calorie restriction. Mech Ageing Dev. 2009; 130:297–307. https://doi.org/10.1016/j.mad.2009.01.004 [PubMed]
- 15. Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977; 252:8731–39. [PubMed]
- 16. Chen Q, Yin G, Stewart S, Hu Y, Lesnefsky EJ. Isolating the segment of the mitochondrial electron transport chain responsible for mitochondrial damage during cardiac ischemia. Biochem Biophys Res Commun. 2010; 397:656–60. https://doi.org/10.1016/j.bbrc.2010.05.137 [PubMed]
- 17. Chen Q, Samidurai A, Thompson J, Hu Y, Das A, Willard B, Lesnefsky EJ. Endoplasmic reticulum stress-mediated mitochondrial dysfunction in aged hearts. Biochim Biophys Acta Mol Basis Dis. 2020; 1866:165899. https://doi.org/10.1016/j.bbadis.2020.165899 [PubMed]
- 18. Barazzoni R, Short KR, Nair KS. Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J Biol Chem. 2000; 275:3343–47. https://doi.org/10.1074/jbc.275.5.3343 [PubMed]
- 19. Choksi KB, Papaconstantinou J. Age-related alterations in oxidatively damaged proteins of mouse heart mitochondrial electron transport chain complexes. Free Radic Biol Med. 2008; 44:1795–805. https://doi.org/10.1016/j.freeradbiomed.2008.01.032 [PubMed]
- 20. Bota DA, Davies KJ. Mitochondrial lon protease in human disease and aging: including an etiologic classification of lon-related diseases and disorders. Free Radic Biol Med. 2016; 100:188–98. https://doi.org/10.1016/j.freeradbiomed.2016.06.031 [PubMed]
- 21. Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013; 4:2308. https://doi.org/10.1038/ncomms3308 [PubMed]
- 22. Liang W, Moyzis AG, Lampert MA, Diao RY, Najor RH, Gustafsson ÅB. Aging is associated with a decline in Atg9b-mediated autophagosome formation and appearance of enlarged mitochondria in the heart. Aging Cell. 2020; 19:e13187. https://doi.org/10.1111/acel.13187 [PubMed]
- 23. Thompson J, Maceyka M, Chen Q. Targeting ER stress and calpain activation to reverse age-dependent mitochondrial damage in the heart. Mech Ageing Dev. 2020; 192:111380. https://doi.org/10.1016/j.mad.2020.111380 [PubMed]
- 24. Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc. 2009; 4:1582–90. https://doi.org/10.1038/nprot.2009.151 [PubMed]
- 25. Paillard M, Csordás G, Huang KT, Várnai P, Joseph SK, Hajnóczky G. MICU1 interacts with the D-ring of the MCU pore to control its Ca2+ flux and sensitivity to Ru360. Mol Cell. 2018; 72:778–85.e3. https://doi.org/10.1016/j.molcel.2018.09.008 [PubMed]
- 26. Chen Q, Thompson J, Hu Y, Das A, Lesnefsky EJ. Metformin attenuates ER stress-induced mitochondrial dysfunction. Transl Res. 2017; 190:40–50. https://doi.org/10.1016/j.trsl.2017.09.003 [PubMed]
- 27. Zhang Y, Ren J. Thapsigargin triggers cardiac contractile dysfunction via NADPH oxidase-mediated mitochondrial dysfunction: role of Akt dephosphorylation. Free Radic Biol Med. 2011; 51:2172–84. https://doi.org/10.1016/j.freeradbiomed.2011.09.005 [PubMed]
- 28. Chen Q, Thompson J, Hu Y, Das A, Lesnefsky EJ. Cardiac specific knockout of p53 decreases ER stress-induced mitochondrial damage. Front Cardiovasc Med. 2019; 6:10. https://doi.org/10.3389/fcvm.2019.00010 [PubMed]
- 29. Xu A, Szczepanek K, Maceyka MW, Ross T, Bowler E, Hu Y, Kenny B, Mehfoud C, Desai PN, Baumgarten CM, Chen Q, Lesnefsky EJ. Transient complex I inhibition at the onset of reperfusion by extracellular acidification decreases cardiac injury. Am J Physiol Cell Physiol. 2014; 306:C1142–53. https://doi.org/10.1152/ajpcell.00241.2013 [PubMed]
- 30. Gundewar S, Calvert JW, Jha S, Toedt-Pingel I, Ji SY, Nunez D, Ramachandran A, Anaya-Cisneros M, Tian R, Lefer DJ. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. 2009; 104:403–11. https://doi.org/10.1161/CIRCRESAHA.108.190918 [PubMed]
- 31. Chen X, Li X, Zhang W, He J, Xu B, Lei B, Wang Z, Cates C, Rousselle T, Li J. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism. 2018; 83:256–70. https://doi.org/10.1016/j.metabol.2018.03.004 [PubMed]
- 32. Zaha VG, Qi D, Su KN, Palmeri M, Lee HY, Hu X, Wu X, Shulman GI, Rabinovitch PS, Russell RR 3rd, Young LH. AMPK is critical for mitochondrial function during reperfusion after myocardial ischemia. J Mol Cell Cardiol. 2016; 91:104–13. https://doi.org/10.1016/j.yjmcc.2015.12.032 [PubMed]
- 33. Mohsin AA, Chen Q, Quan N, Rousselle T, Maceyka MW, Samidurai A, Thompson J, Hu Y, Li J, Lesnefsky EJ. Mitochondrial complex I inhibition by metformin limits reperfusion injury. J Pharmacol Exp Ther. 2019; 369:282–90. https://doi.org/10.1124/jpet.118.254300 [PubMed]
- 34. Duan Q, Song P, Ding Y, Zou MH. Activation of AMP-activated protein kinase by metformin ablates angiotensin II-induced endoplasmic reticulum stress and hypertension in mice in vivo. Br J Pharmacol. 2017; 174:2140–51. https://doi.org/10.1111/bph.13833 [PubMed]
- 35. Simon-Szabó L, Kokas M, Mandl J, Kéri G, Csala M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PLoS One. 2014; 9:e97868. https://doi.org/10.1371/journal.pone.0097868 [PubMed]
- 36. Das A, Durrant D, Koka S, Salloum FN, Xi L, Kukreja RC. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: potential role of attenuated oxidative stress and altered contractile protein expression. J Biol Chem. 2014; 289:4145–60. https://doi.org/10.1074/jbc.M113.521062 [PubMed]
- 37. Samidurai A, Kukreja RC, Das A. Emerging role of mTOR signaling-related miRNAs in cardiovascular diseases. Oxid Med Cell Longev. 2018; 2018:6141902. https://doi.org/10.1155/2018/6141902 [PubMed]
- 38. Brandt C, Nolte H, Henschke S, Engström Ruud L, Awazawa M, Morgan DA, Gabel P, Sprenger HG, Hess ME, Günther S, Langer T, Rahmouni K, Fenselau H, et al. Food perception primes hepatic ER homeostasis via melanocortin-dependent control of mTOR activation. Cell. 2018; 175:1321–35.e20. https://doi.org/10.1016/j.cell.2018.10.015 [PubMed]
- 39. Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. 2015; 33:107–38. https://doi.org/10.1146/annurev-immunol-032414-112116 [PubMed]
- 40. Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO, Hoppel CL. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 1999; 372:399–407. https://doi.org/10.1006/abbi.1999.1508 [PubMed]
- 41. Lesnefsky EJ, Hoppel CL. Ischemia-reperfusion injury in the aged heart: role of mitochondria. Arch Biochem Biophys. 2003; 420:287–97. https://doi.org/10.1016/j.abb.2003.09.046 [PubMed]
- 42. Akande O, Chen Q, Toldo S, Lesnefsky EJ, Quader M. Ischemia and reperfusion injury to mitochondria and cardiac function in donation after circulatory death hearts- an experimental study. PLoS One. 2020; 15:e0243504. https://doi.org/10.1371/journal.pone.0243504 [PubMed]
- 43. Rennison JH, McElfresh TA, Chen X, Anand VR, Hoit BD, Hoppel CL, Chandler MP. Prolonged exposure to high dietary lipids is not associated with lipotoxicity in heart failure. J Mol Cell Cardiol. 2009; 46:883–90. https://doi.org/10.1016/j.yjmcc.2009.02.019 [PubMed]
- 44. Chen Q, Paillard M, Gomez L, Li H, Hu Y, Lesnefsky EJ. Postconditioning modulates ischemia-damaged mitochondria during reperfusion. J Cardiovasc Pharmacol. 2012; 59:101–08. https://doi.org/10.1097/FJC.0b013e31823827cc [PubMed]
- 45. Fernandez-Sanz C, Ruiz-Meana M, Miro-Casas E, Nuñez E, Castellano J, Loureiro M, Barba I, Poncelas M, Rodriguez-Sinovas A, Vázquez J, Garcia-Dorado D. Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 2014; 5:e1573. https://doi.org/10.1038/cddis.2014.526 [PubMed]
- 46. Lesnefsky EJ, Chen Q, Slabe TJ, Stoll MS, Minkler PE, Hassan MO, Tandler B, Hoppel CL. Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: role of cardiolipin. Am J Physiol Heart Circ Physiol. 2004; 287:H258–67. https://doi.org/10.1152/ajpheart.00348.2003 [PubMed]
- 47. Chen Q, Thompson J, Hu Y, Dean J, Lesnefsky EJ. Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion. Am J Physiol Cell Physiol. 2019; 317:C910–21. https://doi.org/10.1152/ajpcell.00190.2019 [PubMed]
- 48. Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda-Saito M, Turkaly PJ, Hoppel CL. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol. 2001; 33:37–47. https://doi.org/10.1006/jmcc.2000.1273 [PubMed]
- 49. Naidoo N. ER and aging-protein folding and the ER stress response. Ageing Res Rev. 2009; 8:150–59. https://doi.org/10.1016/j.arr.2009.03.001 [PubMed]
- 50. Wolfgang MJ, Lane MD. Control of energy homeostasis: role of enzymes and intermediates of fatty acid metabolism in the central nervous system. Annu Rev Nutr. 2006; 26:23–44. https://doi.org/10.1146/annurev.nutr.25.050304.092532 [PubMed]
- 51. Mungrue IN, Pagnon J, Kohannim O, Gargalovic PS, Lusis AJ. CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J Immunol. 2009; 182:466–76. https://doi.org/10.4049/jimmunol.182.1.466 [PubMed]
- 52. Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012; 19:310–20. https://doi.org/10.1038/cdd.2011.98 [PubMed]
- 53. Logue SE, Cleary P, Saveljeva S, Samali A. New directions in ER stress-induced cell death. Apoptosis. 2013; 18:537–46. https://doi.org/10.1007/s10495-013-0818-6 [PubMed]
- 54. Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem. 2012; 151:217–19. https://doi.org/10.1093/jb/mvr143 [PubMed]
- 55. Alam S, Abdullah CS, Aishwarya R, Orr AW, Traylor J, Miriyala S, Panchatcharam M, Pattillo CB, Bhuiyan MS. Sigmar1 regulates endoplasmic reticulum stress-induced C/EBP-homologous protein expression in cardiomyocytes. Biosci Rep. 2017; 37:BSR20170898. https://doi.org/10.1042/BSR20170898 [PubMed]
- 56. Whittington HJ, Hall AR, McLaughlin CP, Hausenloy DJ, Yellon DM, Mocanu MM. Chronic metformin associated cardioprotection against infarction: not just a glucose lowering phenomenon. Cardiovasc Drugs Ther. 2013; 27:5–16. https://doi.org/10.1007/s10557-012-6425-x [PubMed]
- 57. Eurich DT, Weir DL, Majumdar SR, Tsuyuki RT, Johnson JA, Tjosvold L, Vanderloo SE, McAlister FA. Comparative safety and effectiveness of metformin in patients with diabetes mellitus and heart failure: systematic review of observational studies involving 34,000 patients. Circ Heart Fail. 2013; 6:395–402. https://doi.org/10.1161/CIRCHEARTFAILURE.112.000162 [PubMed]
- 58. Holman R. Metformin as first choice in oral diabetes treatment: the UKPDS experience. Journ Annu Diabetol Hotel Dieu. 2007; 13-20. [PubMed]
- 59. Chen Q, Lesnefsky EJ. Metformin and myocardial ischemia and reperfusion injury: moving toward “prime time” human use? Transl Res. 2021; 229:1–4. https://doi.org/10.1016/j.trsl.2020.10.006 [PubMed]
- 60. Lexis CP, van der Horst IC, Lipsic E, Wieringa WG, de Boer RA, van den Heuvel AF, van der Werf HW, Schurer RA, Pundziute G, Tan ES, Nieuwland W, Willemsen HM, Dorhout B, et al, and GIPS-III Investigators. Effect of metformin on left ventricular function after acute myocardial infarction in patients without diabetes: the GIPS-III randomized clinical trial. JAMA. 2014; 311:1526–35. https://doi.org/10.1001/jama.2014.3315 [PubMed]
- 61. Zhuo XZ, Wu Y, Ni YJ, Liu JH, Gong M, Wang XH, Wei F, Wang TZ, Yuan Z, Ma AQ, Song P. Isoproterenol instigates cardiomyocyte apoptosis and heart failure via AMPK inactivation-mediated endoplasmic reticulum stress. Apoptosis. 2013; 18:800–10. https://doi.org/10.1007/s10495-013-0843-5 [PubMed]
- 62. Nam DH, Han JH, Kim S, Shin Y, Lim JH, Choi HC, Woo CH. Activated protein C prevents methylglyoxal-induced endoplasmic reticulum stress and cardiomyocyte apoptosis via regulation of the AMP-activated protein kinase signaling pathway. Biochem Biophys Res Commun. 2016; 480:622–28. https://doi.org/10.1016/j.bbrc.2016.10.106 [PubMed]
- 63. Dong Y, Zhang M, Wang S, Liang B, Zhao Z, Liu C, Wu M, Choi HC, Lyons TJ, Zou MH. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes. 2010; 59:1386–96. https://doi.org/10.2337/db09-1637 [PubMed]
- 64. Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005; 25:9554–75. https://doi.org/10.1128/MCB.25.21.9554-9575.2005 [PubMed]
- 65. Nassif M, Matus S, Castillo K, Hetz C. Amyotrophic lateral sclerosis pathogenesis: A journey through the secretory pathway. Antioxid Redox Signal. 2010; 13:1955–89. https://doi.org/10.1089/ars.2009.2991 [PubMed]
- 66. Basseri S, Lhoták S, Sharma AM, Austin RC. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J Lipid Res. 2009; 50:2486–501. https://doi.org/10.1194/jlr.M900216-JLR200 [PubMed]
- 67. Jian L, Lu Y, Lu S, Lu C. Chemical chaperone 4-phenylbutyric acid reduces cardiac ischemia/reperfusion injury by alleviating endoplasmic reticulum stress and oxidative stress. Med Sci Monit. 2016; 22:5218–27. https://doi.org/10.12659/msm.898623 [PubMed]
- 68. Ghosh AK, Garg SK, Mau T, O’Brien M, Liu J, Yung R. Elevated endoplasmic reticulum stress response contributes to adipose tissue inflammation in aging. J Gerontol A Biol Sci Med Sci. 2015; 70:1320–29. https://doi.org/10.1093/gerona/glu186 [PubMed]
- 69. Varjabedian L, Bourji M, Pourafkari L, Nader ND. Cardioprotection by metformin: beneficial effects beyond glucose reduction. Am J Cardiovasc Drugs. 2018; 18:181–93. https://doi.org/10.1007/s40256-018-0266-3 [PubMed]
- 70. Vial G, Detaille D, Guigas B. Role of mitochondria in the mechanism(s) of action of metformin. Front Endocrinol (Lausanne). 2019; 10:294. https://doi.org/10.3389/fendo.2019.00294 [PubMed]
- 71. Fontaine E. Metformin-induced mitochondrial complex I inhibition: facts, uncertainties, and consequences. Front Endocrinol (Lausanne). 2018; 9:753. https://doi.org/10.3389/fendo.2018.00753 [PubMed]
- 72. Karnewar S, Neeli PK, Panuganti D, Kotagiri S, Mallappa S, Jain N, Jerald MK, Kotamraju S. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: relevance in age-associated vascular dysfunction. Biochim Biophys Acta Mol Basis Dis. 2018; 1864:1115–28. https://doi.org/10.1016/j.bbadis.2018.01.018 [PubMed]
- 73. Wang L, Quan N, Sun W, Chen X, Cates C, Rousselle T, Zhou X, Zhao X, Li J. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc Res. 2018; 114:805–21. https://doi.org/10.1093/cvr/cvy033 [PubMed]
- 74. Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009; 458:1056–60. https://doi.org/10.1038/nature07813 [PubMed]
- 75. Drake JC, Peelor FF 3rd, Biela LM, Watkins MK, Miller RA, Hamilton KL, Miller BF. Assessment of mitochondrial biogenesis and mTORC1 signaling during chronic rapamycin feeding in male and female mice. J Gerontol A Biol Sci Med Sci. 2013; 68:1493–501. https://doi.org/10.1093/gerona/glt047 [PubMed]
- 76. Wang S, Kandadi MR, Ren J. Double knockout of Akt2 and AMPK predisposes cardiac aging without affecting lifespan: role of autophagy and mitophagy. Biochim Biophys Acta Mol Basis Dis. 2019; 1865:1865–75. https://doi.org/10.1016/j.bbadis.2018.08.011 [PubMed]
- 77. Li Y, Chen Y. AMPK and autophagy. Adv Exp Med Biol. 2019; 1206:85–108. https://doi.org/10.1007/978-981-15-0602-4_4 [PubMed]
- 78. Turdi S, Fan X, Li J, Zhao J, Huff AF, Du M, Ren J. AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell. 2010; 9:592–606. https://doi.org/10.1111/j.1474-9726.2010.00586.x [PubMed]
- 79. Zhao D, Yang J, Yang L. Insights for oxidative stress and mTOR signaling in myocardial ischemia/reperfusion injury under diabetes. Oxid Med Cell Longev. 2017; 2017:6437467. https://doi.org/10.1155/2017/6437467 [PubMed]
- 80. Morita M, Prudent J, Basu K, Goyon V, Katsumura S, Hulea L, Pearl D, Siddiqui N, Strack S, McGuirk S, St-Pierre J, Larsson O, Topisirovic I, et al. mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol Cell. 2017; 67:922–35.e5. https://doi.org/10.1016/j.molcel.2017.08.013 [PubMed]
- 81. Flynn JM, O’Leary MN, Zambataro CA, Academia EC, Presley MP, Garrett BJ, Zykovich A, Mooney SD, Strong R, Rosen CJ, Kapahi P, Nelson MD, Kennedy BK, Melov S. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013; 12:851–62. https://doi.org/10.1111/acel.12109 [PubMed]
- 82. Chiao YA, Kolwicz SC, Basisty N, Gagnidze A, Zhang J, Gu H, Djukovic D, Beyer RP, Raftery D, MacCoss M, Tian R, Rabinovitch PS. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging (Albany NY). 2016; 8:314–27. https://doi.org/10.18632/aging.100881 [PubMed]
- 83. Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, Watkins M, Shavlakadze T, Zhou W, Quinn D, Glass DJ, Klickstein LB. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med. 2018; 10:eaaq1564. https://doi.org/10.1126/scitranslmed.aaq1564 [PubMed]
- 84. Budanov AV, Karin M. P53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008; 134:451–60. https://doi.org/10.1016/j.cell.2008.06.028 [PubMed]
- 85. Morrison A, Chen L, Wang J, Zhang M, Yang H, Ma Y, Budanov A, Lee JH, Karin M, Li J. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 2015; 29:408–17. https://doi.org/10.1096/fj.14-258814 [PubMed]
- 86. Quan N, Sun W, Wang L, Chen X, Bogan JS, Zhou X, Cates C, Liu Q, Zheng Y, Li J. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. FASEB J. 2017; 31:4153–67. https://doi.org/10.1096/fj.201700063R [PubMed]
- 87. Alexander A, Walker CL. The role of LKB1 and AMPK in cellular responses to stress and damage. FEBS Lett. 2011; 585:952–57. https://doi.org/10.1016/j.febslet.2011.03.010 [PubMed]
- 88. Maiuri MC, Kroemer G. Autophagy in stress and disease. Cell Death Differ. 2015; 22:365–66. https://doi.org/10.1038/cdd.2014.236 [PubMed]
- 89. Ro SH, Xue X, Ramakrishnan SK, Cho CS, Namkoong S, Jang I, Semple IA, Ho A, Park HW, Shah YM, Lee JH. Tumor suppressive role of sestrin2 during colitis and colon carcinogenesis. Elife. 2016; 5:e12204. https://doi.org/10.7554/eLife.12204 [PubMed]
- 90. Pasha M, Eid AH, Eid AA, Gorin Y, Munusamy S. Sestrin2 as a novel biomarker and therapeutic target for various diseases. Oxid Med Cell Longev. 2017; 2017:3296294. https://doi.org/10.1155/2017/3296294 [PubMed]
- 91. Babiker F, Al-Jarallah A, Al-Awadi M. Effects of cardiac hypertrophy, diabetes, aging, and pregnancy on the cardioprotective effects of postconditioning in male and female rats. Cardiol Res Pract. 2019; 2019:3403959. https://doi.org/10.1155/2019/3403959 [PubMed]
- 92. Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003; 93:292–301. https://doi.org/10.1161/01.RES.0000087542.26971.D4 [PubMed]
- 93. Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M, Sadoshima J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2α/activating transcription factor 4 pathway. Circ Res. 2013; 113:1253–64. https://doi.org/10.1161/CIRCRESAHA.113.301787 [PubMed]
- 94. Tanaka-Esposito C, Chen Q, Lesnefsky EJ. Blockade of electron transport before ischemia protects mitochondria and decreases myocardial injury during reperfusion in aged rat hearts. Transl Res. 2012; 160:207–16. https://doi.org/10.1016/j.trsl.2012.01.024 [PubMed]
- 95. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014; 515:431–35. https://doi.org/10.1038/nature13909 [PubMed]
- 96. Downey JM, Cohen MV. Why do we still not have cardioprotective drugs? Circ J. 2009; 73:1171–77. https://doi.org/10.1253/circj.cj-09-0338 [PubMed]
- 97. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007; 357:1121–35. https://doi.org/10.1056/NEJMra071667 [PubMed]
- 98. Li YH, Wang MT, Huang WC, Hwang JJ. Management of acute coronary syndrome in patients with suspected or confirmed coronavirus disease 2019: consensus from Taiwan society of cardiology. J Formos Med Assoc. 2021; 120:78–82. https://doi.org/10.1016/j.jfma.2020.07.017 [PubMed]
- 99. Zhang W, Zhang H, Yao W, Li L, Niu P, Huo Y, Tan W. Morphometric, hemodynamic, and multi-omics analyses in heart failure rats with preserved ejection fraction. Int J Mol Sci. 2020; 21:3362. https://doi.org/10.3390/ijms21093362 [PubMed]
- 100. Sabbah HN. Targeting the mitochondria in heart failure: a translational perspective. JACC Basic Transl Sci. 2020; 5:88–106. https://doi.org/10.1016/j.jacbts.2019.07.009 [PubMed]
- 101. Bocchi EA, Avila MS, Ayub-Ferreira SM. Aging, cardiotoxicity, and chemotherapy. Aging (Albany NY). 2019; 11:295–96. https://doi.org/10.18632/aging.101776 [PubMed]
- 102. Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ, Larner AC. Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem. 2011; 286:29610–20. https://doi.org/10.1074/jbc.M111.226209 [PubMed]
- 103. Paillard M, Gomez L, Augeul L, Loufouat J, Lesnefsky EJ, Ovize M. Postconditioning inhibits mPTP opening independent of oxidative phosphorylation and membrane potential. J Mol Cell Cardiol. 2009; 46:902–09. https://doi.org/10.1016/j.yjmcc.2009.02.017 [PubMed]
- 104. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003; 278:36027–31. https://doi.org/10.1074/jbc.M304854200 [PubMed]
- 105. Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, Lesnefsky EJ. Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem Biophys Res Commun. 2011; 415:533–38. https://doi.org/10.1016/j.bbrc.2011.10.037 [PubMed]
- 106. Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 2006; 319:1405–12. https://doi.org/10.1124/jpet.106.110262 [PubMed]
- 107. Steel R, Torrie J. Principles and procedures of statistics. Mc Graw-Hill, New York, 1960.