



Introduction
Parkinson’s disease (PD) is a main neurodegenerative diseases (ND), in that degeneration of the nervous system is marked by a loss of dopamine in the substantia nigra pars compacta (SNPC). This disease is localized in the brain and/or in the spinal cord, patients remain without any symptoms for a considerable duration [1, 2]. PD causes are still unknown but the presence of Lewy bodies (clumps of a-synuclein and ubiquitin proteins in neurons) was shown from the early steps of the condition. PD shows tremor symptoms, instability in postural, rigidity and postural instability. These symptoms appear only when majority of the dopaminergic (DAergic) cells has been lost in the SNPC, meaning that the smooth, which are the regulation control of striatal motor circuits, was also lost [3]. Non-motor symptoms, including depression and rapid eye movement (REM)-associated sleep behavior disorder (RBD), could involve the initiation of PD.
Aging is a main risk marker of neurodegeneration, as it may dysregulate the different signaling that modulate homeostasis processes in cells. Cells with neurodegeneration are the sites of numerous molecular and cellular dysregulation [4]. Numerous metabolic processes, including inflammation and oxidative stress (OS), could involve to neurodegenerative mechanisms. PD highlights a metabolic reprogramming involving stimulation of OS and inflammation [5, 6]. For a few years now, the WNT/β-catenin pathway was shown to be a major signaling systems implicated in PD [7, 8] and its dysregulation an early sign in the development of the condition [9].
Currently, drug therapies the main efficient and widely utilized treatments in PD are the use of levodopa, DA agonists, amantadine, monoamine oxidase B (MAO-B) inhibitors [10], catechol-O-methyltransferase (COMT) negative regulators [11], and many anticholinergic therapies. As physiotherapy, the nuclear destruction and stimulation of deep brain [12] are novel strategies, showing a great interest. Moreover, adjuvant therapies also are interesting for re- mission and preliminary therapy in PD. Although these drugs can counteracted many symptoms of PD to some extent, these cannot counteract PD development and can lead to many adverse effects. Currently, cannabidiol (CBD) is one of the main interesting therapy way for NDs [13, 14].
Cannabidiol (CBD) is a non-psychotomimetic phytocannabinoid derived from the Cannabis sativa plant. The plant possesses many therapeutic properties for a range of neurodegenerative diseases [13–15] and, in the few years, CBD has presented increasing interest as a possible anxiolytic therapy [16–18]. CBD decreases the stimulation of GSK3-β, an negative modulator of the WNT/β-catenin pathway [19], and has been found to suppress inflammatory signaling [20, 21] and oxidative stress [22]. The present review focuses on these metabolic mechanisms and the potential beneficial effects of cannabidiol (CBD) as part of a therapeutic strategy in PD.
Parkinson’s disease and oxidative stress
Several findings have documented the stimulation of OS in PD [23]. Mitochondrial deregulation was shown in PD by increasing energy production and then, the release of reactive oxygen species (ROS) [24]. A decrease in mitochondrial activity involves cell damage and death through a decrease in energy production due to the enhancement of OS [5, 25]. OS and mitochondrial depletion have been found to be correlated with dementia and cell death [26–28]. A decrease in in the activity of the respiratory chain in the SNPC of a patient with PD is correlated with an augmentation in ROS production and apoptosis initiation [24, 29, 30].
Body can produce free radicals of oxygen for oxidative metabolism. In the aerobic respiration, molecular oxygen (O2) is diminished to water molecules in mitochondria. Through this phenomenon, O2, H2O2 and OH are generated by a leakage of oxygen [6]. Phagocytic cells, in response to inflammation and infection, produce high rates of NO, O2 and H202 to protect the human body and thereby diminish this infection. However, the radicals generated could damage cells [31].
Many enzymes, such as monoamine oxidase (MAO), L-amino acid oxidase and tyrosine hydroxylase, are implicated in metabolism of dopamine and in ROS production [32]. ROS production is also generated by inflammation. However, several types of signaling activity can act together with ROS production. The ROS-induced proteins aggregation could lead to inflammatory process in microglia [33]. Four processes enhanced in PD are associated with inflammation and OS: stimulation in iron rates, the diminution in glutathione (GSH) rates, the decrease of 26S proteasomal function and the deregulation of mitochondrial complex I regulation [34, 35]. During the physiologic stage, MAO generates H2O2, but during PD development, H2O2 is changed into hydroxyl radicals (OH) through iron by the Fenton reaction. Then, H2O2 and OH enhance OS [36]. In the PD cytosol, H2O2 and OH oxidized GSH [37], involving leakage of GSH. The GSH leakage generates the transformation of glutamate and cysteine into peptides called glutamyl and cysteinyl. These peptides have a adverse effect on dopaminergic cells by linking the membrane of cells and by increasing ROS production in dopaminergic neurons. They also diminish the stimulation of the mitochondria complex I, which leads to OS and ROS production [38]. DAergic cells are not available to bind misfolded proteins because of the impair in proteasomal mechanisms [39]. OS involves the carbonylation of proteins, leading to an unrepairable and irreversible change. Carbonylation is a phenotype of senescence of cells enhancing the aggregation of proteins. In PD, proteins aggregation is a main pathological feature of nigrostriatal DAergic neurons. Proteins aggregation leads to neuroinflammation and OS [40].
Parkinson’s disease and inflammation
Recent PD studies have presented that inflammation has a main action [41] by activating the apoptosis pathways in dopaminergic cells [42, 43]. The association between inflammation and PD is 2-way; inflammatory processes enhance dopaminergic cells death, death of DAergic cells, in a vicious loop, can also stimulate inflammation [44]. Furthermore, inflammatory markers lead to OS, involving DAergic cells to stimulate death signaling [45]. In PD, many inflammatory markers, such as microglia, show a major action [46]. Microglia stimulation can activate their pro-inflammatory enzymes (including inducible nitric oxide synthase and COX) and releasing of inflammatory cytokines (including tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), C-X-C motif chemokine ligand 12 (CXCL12), interleukin (IL)-6 and IL-1β) [47]. In microglia, the NF-κB pathway plays a main action in the generation of these inflammatory cytokines [48]. TNF-α stimulates apoptosis through the TNF-R1 receptor death domain which activates the caspases 1 and 3 [49]. TNF-α decreases c-Rel–NF-κB. c-Rel–NF-κB plays a neuroprotective function by inhibiting apoptosis via the B-cell lymphoma-extra-large signaling in DAergic neurons [48]. PD shows increased rates of CXCR4 (named fusin) expression and its ligand CXCL12. The dimer composed by CXCR4-CXCL12 stimulates caspase 3, enhancing apoptosis and then neural cell death [50, 51]. The dimer of IFN-γ–IFNGR pathway leads to the phosphorylation of the leucine-rich repeat kinase 2 (LRRK2) protein [52]. In microglia and in DAergic neurons, LRRK2 binds to several cellular mechanisms. Stimulated LRRK2 downregulates expression of c-Rel–NF-κB. Then, inflammatory process is stimulated by decreasing c-Rel–NF-κB activity [53, 54]. LRRK2 stimulation can lead to the initiation of tau oligomers, stimulating cell death signaling [55, 56]. In cells, LRRK2 modulates several vesicle trafficking and its up-regulation is correlated with an increase in inflammatory cytokines [57].
WNT/β-catenin pathway
The WNT name is comes from “Wingless drosophila melanogaster” and its mouse homolog “Int”. The WNT/β-catenin pathway is implicated in many signals and molecular processes, including cell proliferation, embryogenesis, cell migration and cell polarity, apoptosis, and organogenesis [58]. Nevertheless, the WNT/β-catenin pathway can be deregulated during numerous pathological states, such as inflammation, neurological disorders, metabolic diseases, tissue fibrosis and cancer processes [59].
The WNT pathway belongs to the family of secreted lipid-modified glycoproteins [60]. WNT ligands are secreted by both immune cells and neurons located in the CNS [61]. Modulation of the WNT/β-catenin pathway implicates metabolic pathways, embryony development, cell fate, and epithelial-mesenchymal transition (EMT). WNT/β-catenin pathway deregulation leads to numerous NDs, such as PD [6, 62–64]. At the transcriptional level, WNT signaling is primarily mediated by a family of transcription factors known as the β-catenin/T-cell factor/lymphoid enhancer factor (TCF/LEF). The cytoplasmic accumulation of β-catenin is generated by the complex “AXIN, tumor suppressor adenomatous polyposis coli (APC), and glycogen synthase kinase-3 (GSK-3β)”. In WNT ligands absence, this complex enhances to phosphorylate cytoplasmic β-catenin and involves its proteasomal degradation. In presence of the WNT ligands, β-catenin binds to Frizzled (FZL) and LDL receptor-related protein 5/6 (LRP 5/6), thereby stopping the complex and preventing β-catenin proteasomal degradation. β-catenin translocates to the nucleus to bind with TCF/LEF. This in turn activates WNT target genes [65–67].
GSK-3β is a major negative modulator of the WNT/β-catenin pathway [68–73]. As an intracellular serine-threonine kinase, GSK-3β is a controller of the WNT/β-catenin pathway [74]. GSK-3β is implicated in the modulation of numerous pathophysiological pathways, including cell membrane pathway, cell polarity, and inflammatory process [75–77]. GSK-3β acts by downregulating cytoplasmic β-catenin and stabilizes it to activate its nuclear translocation. Inflammatory process is an age-related mechanism correlated with the activation of GSK-3β activity and the decrease of the WNT/β-catenin pathway [78] (Figure 1).
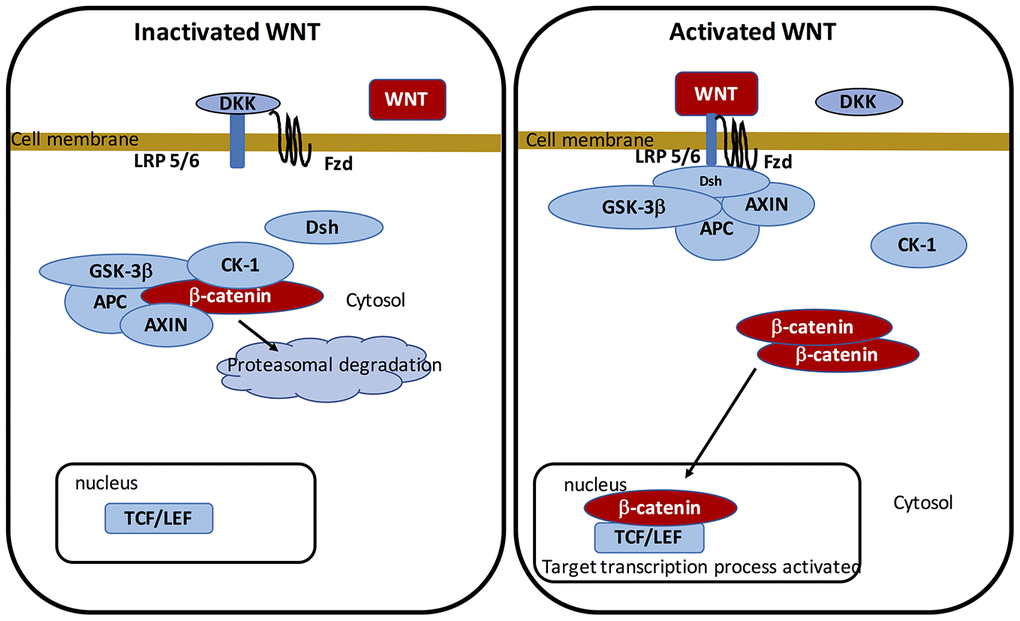
Figure 1. On-state and off-state activation of the WNT pathway.
Parkinson’s disease and WNT/β-catenin pathway
Dysregulation in the WNT/β-catenin pathway is intricately involved in the development of PD [7, 8]. WNT/β-catenin pathway deregulation is an initiating event in PD [9]. Numerous cell biological roles damaged in PD are modulated by the WNT/β-catenin pathway, including axonal function, microtubule stability and membrane trafficking [79, 80]. The mesencephalic dopaminergic neuron-astrocyte interplay is modulated by WNT-1 controlled Frizzled-1/β-catenin pathway [81]. In normal conditions, LRRK2 binds to the WNT/β-catenin pathway and Dishevelled (DSH) proteins to downregulate the β-catenin destruction complex and to increase the WNT/β-catenin pathway [9]. In majority, PD is an idiopathic disease. Nevertheless, in familial Parkinsonism, genes are generally associated with PARK genes. PARKs mutations, codifying for LRRK2, have been observed to be an etiology of PD family forms [82]. LRRK2 mutations decrease the LRRK2-LRP5/6 binding affinity and are associated with a decrease in WNT pathway activity [83]. PARK2 gene encodes the E3 ubiquitin ligase Parkin. Parkin genetic damages are involved in PD initiation and operate as β-catenin inhibitors leading to β-catenin ubiquitination and proteasomal degradation [8]. In PD, DKK1 and GSK-3β are increased [84]. PD mice models present an interplay between inflammation, OS and the WNT/β-catenin signaling [85].
Oxidative stress and WNT/β-catenin pathway
FoxO (Forkhead box class O) transcription factors are important intracellular modulators of numerous molecular pathways, such as glucose production and cell response to OS [86]. ROS production is correlated with the diminution of the WNT/β-catenin pathway by diverting β-catenin from TCF/LEF to FoxO [87]. This leads to the accumulation and binding of β-catenin to FoxO as a co-regulator, and in stimulating FoxO nuclear transcriptional activity [88, 89]. FoxO stimulates apoptotic genes expression [90–92]. FoxO3a interrupts the cell-cycle by activating the production of the cyclin-dependent kinase inhibitor p27 kip1 and the diminution of expression of cyclin D1 [93, 94]. The stimulation of FoxO induces of apoptosis [95]. Nevertheless, the increase of the WNT/β-catenin pathway can decrease FoxO3a in the cytoplasm to counteract mitochondrial membrane permeability loss, release of cytochrome c, phosphorylation of Bad, and the stimulation of caspases. Stimulation of the WNT/β-catenin pathway also activates OS and ROS production [96].
Inflammation and WNT/β-catenin pathway
The activation of the WNT/β-catenin pathway decreases inflammation and enhances neuroprotection through several interplays between microglia/macrophages and astrocytes [81, 97].
Numerous findings have observed an interplay between the WNT/β-catenin and NF-κB pathways, major markers of inflammatory process [98]. The NF-κB transcription factor family comprises 5 compounds in the cytoplasm under unactuated steps: NF-κB 1 (p50/p105), NF-κB 2 (p52/p100), RelA (p65), RelB and c-Rel [99]. β-catenin complexes with RelA and p50 to decrease NF-κB signaling activity [100]. Furthermore, by binding to the PI3K, β-catenin decreases the activity of NF-κB pathway [101]. The downregulatory role of β-catenin on NF-κB pathway has been shown in several cellular signaling processes, including fibroblasts, epithelial cells, hepatocytes and osteoblasts [98]. Moreover, GSK-3β stimulation activity inhibits the β-catenin and then, a stimulation of NF-κB pathway [102]. The possible protective role of β-catenin is caused by the stimulation of the PI3K/Akt pathway and the decrease in the TLR4-driven inflammatory response [103]. NF-κB pathway stimulation inhibits the β-catenin/TCF/LEF complex by increasing LZTS2 [104]. DKK1, a negative modulator of the WNT pathway, is a target gene of the NF-κB pathway involving a negative interplay decreasing the β-catenin pathway [105]. Stimulated β-catenin downregulates the NF-κB-mediated transcription of pro-inflammatory genes. This phenomenon is directly modulated by the activity of GSK-3β [106, 107].
Cannabidiol
Cannabinoids provide from a heterogeneous group of components characterized by 3 major components: endogenous, synthetic and phytocannabinoids [108, 109]. CBD is a non-psychotomimetic phytocannabinoid derived from the Cannabis sativa plant. The Cannabis sativa plant generates more than sixty-six components such as delta9-tetrahydrocannabinol (THC), causing psychological effects, and CBD, the main non-psychotomimetic component in the Cannabis sativa plant [110]. CBD presents no interaction with blood pressure or body temperature and no association with psychomotor psychological functions such as THC [111]. CBD attenuates damages in brain correlated with neurodegenerative processes. Human bodies could tolerate high doses of CBD [111]. Furthermore, CBD can interact with synaptic plasticity and induce neurogenesis mechanism. The mechanisms of CBD effects remain unclear but seem to have several pharmacological targets. Traditional medicines used Cannabis sativa for centuries. CBD, a major components of Cannabis sativa, has recently presented considerable interest for its potential role with respect to many neuropsychiatric disorders [112]. CBD presents a large spectrum of possible therapeutic properties, including anxiolytic, antidepressant, neuroprotective, anti-inflammatory and immunomodulatory processes [109]. Cannabinoids are a novel class of drugs due to their possible role in treating neuropsychiatric diseases [13], including schizophrenia, epilepsy, addiction and neonatal hypoxic-ischemic encephalopathy [113]. In schizophrenia, CBD stimulates the WNT/β-catenin and PI3K/Akt pathways to lead to therapy actions [14, 114, 115].
OS contributes to neurodegeneration. Thus, neuron cells present a functional or sensory loss in neurodegeneration. For life, even if oxygen is needed, an unbalanced metabolic process and an increased production of reactive oxygen species ends up in several NDs, including AD and PD. Free radicals lead to damages in protein and DNA, stimulate inflammation processes and apoptosis [116]. Some findings have shown that secondary plant metabolites, from medicinal herbs, could show lead components for medication production for inflammation and OS therapy, leading to protect from loss in neuronal cell [117]. Among them, CBD could be a prototype for anti-inflammatory and antioxidative therapy for these diseases where inflammation and OS have major actions in their etiologies and initiation [118].
In the CNS, CBD has been shown to possess anti-inflammatory actions, thus being useful for neuro-inflammatory diseases [119], and therapy of spasticity and pain [120]. Based on its anticonvulsant roles, CBD has been used as a therapy for epilepsy [121], and also for the therapy for sleep disorders [122] due to its capability to control serotonin transmission [123]. CBD possesses interesting roles for psychiatric disorders, such as schizophrenia [124], but it also presents other possible actions, such as anxiolytic and antidepressant roles [125, 126]. The neuroprotective action of CBD for the management of certain other NDs has also been investigated in different studies that have yielded many positive results [13].
Parkinson’s disease and cannabidiol
Recent clinical investigations have presented the interest of using CBD for its antiparkinsonian properties [127–131]. CBD can significantly reduce 6-OHDA-induced neurotoxic actions in mice, and this neuro-protective role could be controlled through cannabinoid receptor-independent anti-inflammatory and antioxidant actions [127]. CBD can also target and reduce the different inflammatory factors, including COX-2 and NF-κB. These factors have been found to be blocked by the CBD effect on PPARγ receptors [131, 132]. Moreover, CBD can reduce DA depletion and slow down the increase in OS [13, 133]. The latter evidence suggests that CBD has antioxidant properties and can diminish the nigrostriatal dopaminergic neurodegeneration fibers observed in PD [134]. Furthermore, CBD presents a high possible antioxidant actions, compared to ascorbate, for cortical neurons treated with toxic glutamate concentrations [15]. The neuroprotective action was shown regardless of whether the insult was due to the stimulation of N-methyl-D-aspartate (NMDA) receptor, a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, or kainate receptors and, it is not controlled by CB receptors since the CB antagonist is not damaged [135]. The recent result may present that CBD could be a possible antioxidant without psychotropic adverse effects, directly controlled by CB receptors.
Stimulation of the WNT/β-catenin pathway by cannabidiol: a possible therapeutic strategy (Figure 2)
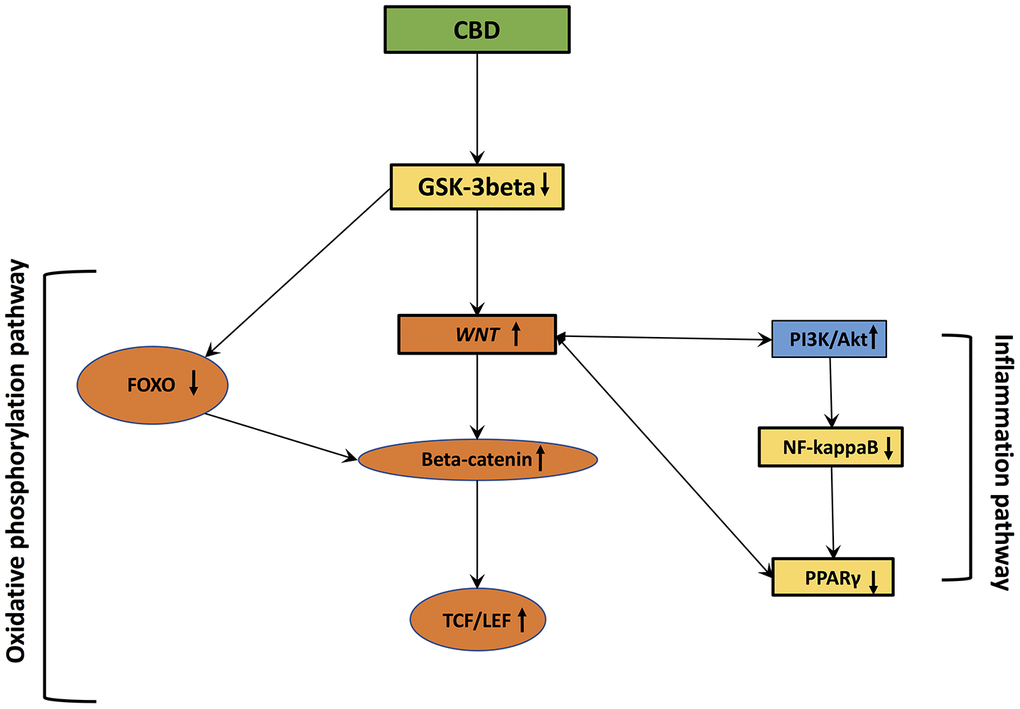
Figure 2. Cannabidiol interactions with oxidative stress and inflammation.
A study have observed that mutant OCD mouse models presented stimulated GSK-3β activity, suggesting that GSK-3β downregulation can provide a therapy for perseverative behavior [136].
Dysfunction of GSK-3β is implicated in the pathogenesis of several disorders, such as neuropsychiatric disorders [137]. GSK3β is known to be the main inhibitor of the WNT/β-catenin pathway [72, 138–140]. GSK-3β downregulates the WNT/β-catenin pathway by inhibiting β-catenin cytoplasmic stabilization and its nuclear translocation [83]. Moreover, many findings have observed a link between neuro-inflammation and the augmentation of the GSK-3β and a decrease in the activity of the WNT/β-catenin pathway and the Akt pathway [68].
CBD downregulates expression of GSK-3β through the promotion of PI3K/Akt signaling [141, 142]. PI3K/Akt signaling regulates GSK-3β activity [143]. Cannabinoids control the PI3K/Akt/GSK-3β axis [144, 145]. Gene coding for the PI3K/Akt pathway is increased in CBD-GMSCs (mesenchymal stem cells derived from gingiva treated by CBD) [141]. Diminution of β-catenin activity reduces the expression of PI3K/Akt pathway [146]. In schizophrenia, CBD stimulates the WNT/β-catenin and PI3K/Akt pathways to enhance therapeutic actions [13]. Cannabinoids can directly control the PI3K/Akt/GSK-3β axis [144, 145]. In GMSCs treated by CBD, genes coding for the PI3K/Akt signaling are increased [141]. CBD downregulates GSK-3β activity by stimulating PI3K/Akt pathway [141]. In neurons and glial cells, CBD can stimulate the PI3K/Akt pathway by interacting with CB1 receptor and, in a less manner in the immune system with CB2 receptor [147, 148].
CBD can reduce the activity of the pro-inflammatory factors COX-2 and NF-κB. These effects are stopped by the combination of CBD and PPARγ receptors. The neuroprotective effects of CBD are generated by anti-inflammatory actions modulated by both CB1 and PPARγ [149]. By interacting with PPARγ, CBD can activate the canonical WNT pathway to reduce inflammation and OS [14].
Oxidative stress and cannabidiol
The energy production and metabolism of glucose implicated in OS are regulated by the FOXO transcription factors [86]. This relationship between β-catenin and FOXO leads to the promotion of cell quiescence and cell cycle stop. β-catenin interrupts its complex with TCF/LEF by binding with FOXO [87]. β-catenin did not lead to nuclear translocation to and accumulates in the cytoplasm to inactivate the WNT/β-catenin pathway [88, 89].
CBD can reduce the redox balance through the modification of both the rate and activity of oxidants and antioxidants [22]. CBD stops free radical chain reactions through the capture of free radicals and then by reducing their activities [150]. CBD downregulates the oxidative conditions through the prevention of the initiation of superoxide radicals, produced by xanthine oxidase (XO) and NADPH oxidase (NOX1 and NOX4) [151, 152]. Moreover, CBD can enhance the diminution in NO levels [153]. CBD also diminishes ROS production through the chelation of the transition metal ions implicated in the Fenton reaction to enhance hydroxyl radicals [154]. CBD acts on the antioxidant BHT (butylated hydroxytoluene) to prevent dihydrorodamine oxidation in the Fenton reaction [15].
The antioxidant activity of CBD is characterized by the stimulation of redox-sensitive transcription factor associated with the Nrf2 (Nuclear factor-erythroid 2 related factor 2) [155], which controls the transcription of cytoprotective genes [156]. Superoxide dismutase (SOD) and the enzymatic activities of Cu, Zn and Mn-SOD, controlling superoxide radicals metabolic processes, are increased by CBD [157]. Glutathione peroxidase and reductase are also increased by CBD, decreasing the malonaldehyde (MDA) levels [158]. Enzymatic activities are altered during oxidative modifications of proteins. CBD, by targeting glutathione and cytochrome P450, can inhibit their biological activity and thus decrease oxidative stress [153, 159]. Moreover, through the decrease in ROS levels, CBD can prevent and protect non-enzymatic antioxidants [157], including vitamins A, E and C [160].
Inflammation and cannabidiol
Cannabinoids present anti-inflammatory action by endogenous receptors, including CB1 and CB2 [161]. N-Oleoyl glycine (OLGly), a lipoamino acid, activates adipogenic genes including PPARγ, a marker of inflammation, and the expression of mRNA of the CB1 receptor. Inhibition of the CB1 receptor by SR141716 downregulates the actions of OLGly on PPARγ. Moreover, OLGly activates the Akt pathway to inhibit FoxO activity [162]. CBD can bind PPARγ [14, 163]. PPARγ is a major factor of inflammation through its interaction with NFκB. This binding acts on the ligand-binding domain of PPARγ and the Rel homology domain region of the p65 subunit of NFκB. Proteasomal degradation of p65 is caused by the Lys48-linked polyubiquitin of the ligand-binding domain of PPARγ [164]. Thus, PPARγ can modulate inflammation through the ubiquitination proteasomal degradation of p65 leading to the control of cyclooxygenase (COX2), TNF-α, IL-1β and IL-6 [14]. PPARs are ligand-activated transcription factors binding PPREs (PPAR-response elements), and are implicated in several dysregulated mechanisms, including cell differentiation, protein metabolisms, lipid metabolisms, carcinogenesis [165, 166], adipocyte differentiation, insulin sensitivity and inflammation [167, 168]. PPARγ ligands, including thiazolidinediones (TZDs), can diminish inflammation [169]. A negative crosstalk between PPARγ and the WNT/β-catenin pathway has been well documented [138, 170–172]. The PI3K/Akt pathway, enhancing by β-catenin [140, 171, 173–175], interacts through the phosphorylation of GSK-3β to decrease PPARγ [176]. PPARγ agonists inhibit β-catenin through the stimulation of GSK-3β activity [177]. PPARγ agonists activate DKK1 expression to decrease the WNT/β-catenin pathway [178]. Moreover, PPARγ agonists stimulate GSK-3β activity to inhibit β-catenin [177]. In parallel, β-catenin directly inhibits NFκB activity [106, 107].
Conclusions
To date, few studies have studied CBD as a potential alternative therapeutic solution in treating PD. However, CBD is attracting increasing interest in this context because of its possible inhibitory effect on OS and inflammation and the fact that, at low doses there are few side effects. WNT/β-catenin pathway activity is diminished in the development of PD. By stimulating the WNT/β-catenin pathway, through the decrease of GSK-3β, CBD may be an integral part of an innovative therapeutic treatment of the condition. Future investigated studies should therefore focus on CBD and its many relationships with the development and treatment of PD.
Author Contributions
All authors listed have contributed to this work and approved its submission for possible publication.
Acknowledgments
The authors thank Brian Keogh, PhD, for English correction and proof reading.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
References
- 1. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004; 318:121–34. https://doi.org/10.1007/s00441-004-0956-9 [PubMed]
- 2. Grinberg LT, Rueb U, Alho AT, Heinsen H. Brainstem pathology and non-motor symptoms in PD. J Neurol Sci. 2010; 289:81–88. https://doi.org/10.1016/j.jns.2009.08.021 [PubMed]
- 3. Maguire-Zeiss KA, Federoff HJ. Future directions for immune modulation in neurodegenerative disorders: focus on Parkinson’s disease. J Neural Transm (Vienna). 2010; 117:1019–25. https://doi.org/10.1007/s00702-010-0431-6 [PubMed]
- 4. Yin F, Boveris A, Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal. 2014; 20:353–71. https://doi.org/10.1089/ars.2012.4774 [PubMed]
- 5. Kim GH, Kim JE, Rhie SJ, Yoon S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp Neurobiol. 2015; 24:325–40. https://doi.org/10.5607/en.2015.24.4.325 [PubMed]
- 6. Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Thermodynamics in Neurodegenerative Diseases: Interplay Between Canonical WNT/Beta-Catenin Pathway-PPAR Gamma, Energy Metabolism and Circadian Rhythms. Neuromolecular Med. 2018; 20:174–204. https://doi.org/10.1007/s12017-018-8486-x [PubMed]
- 7. Parish CL, Castelo-Branco G, Rawal N, Tonnesen J, Sorensen AT, Salto C, Kokaia M, Lindvall O, Arenas E. Wnt5a-treated midbrain neural stem cells improve dopamine cell replacement therapy in parkinsonian mice. J Clin Invest. 2008; 118:149–60. https://doi.org/10.1172/JCI32273 [PubMed]
- 8. Rawal N, Corti O, Sacchetti P, Ardilla-Osorio H, Sehat B, Brice A, Arenas E. Parkin protects dopaminergic neurons from excessive Wnt/beta-catenin signaling. Biochem Biophys Res Commun. 2009; 388:473–78. https://doi.org/10.1016/j.bbrc.2009.07.014 [PubMed]
- 9. Berwick DC, Harvey K. The importance of Wnt signalling for neurodegeneration in Parkinson’s disease. Biochem Soc Trans. 2012; 40:1123–28. https://doi.org/10.1042/BST20120122 [PubMed]
- 10. Alborghetti M, Nicoletti F. Different Generations of Type-B Monoamine Oxidase Inhibitors in Parkinson’s Disease: From Bench to Bedside. Curr Neuropharmacol. 2019; 17:861–73. https://doi.org/10.2174/1570159X16666180830100754 [PubMed]
- 11. Müller T. Pharmacokinetics and pharmacodynamics of levodopa/carbidopa cotherapies for Parkinson’s disease. Expert Opin Drug Metab Toxicol. 2020; 16:403–14. https://doi.org/10.1080/17425255.2020.1750596 [PubMed]
- 12. Parmar M, Grealish S, Henchcliffe C. The future of stem cell therapies for Parkinson disease. Nat Rev Neurosci. 2020; 21:103–15. https://doi.org/10.1038/s41583-019-0257-7 [PubMed]
- 13. Fernández-Ruiz J, Sagredo O, Pazos MR, García C, Pertwee R, Mechoulam R, Martínez-Orgado J. Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid? Br J Clin Pharmacol. 2013; 75:323–33. https://doi.org/10.1111/j.1365-2125.2012.04341.x [PubMed]
- 14. Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Effects of cannabidiol interactions with Wnt/β-catenin pathway and PPARγ on oxidative stress and neuroinflammation in Alzheimer’s disease. Acta Biochim Biophys Sin (Shanghai). 2017; 49:853–66. https://doi.org/10.1093/abbs/gmx073 [PubMed]
- 15. Campos AC, Fogaça MV, Sonego AB, Guimarães FS. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol Res. 2016; 112:119–27. https://doi.org/10.1016/j.phrs.2016.01.033 [PubMed]
- 16. Schier AR, Ribeiro NP, Silva AC, Hallak JE, Crippa JA, Nardi AE, Zuardi AW. Cannabidiol, a Cannabis sativa constituent, as an anxiolytic drug. Braz J Psychiatry. 2012 (Suppl 1); 34:S104–10. https://doi.org/10.1590/S1516-44462012000500008 [PubMed]
- 17. Micale V, Di Marzo V, Sulcova A, Wotjak CT, Drago F. Endocannabinoid system and mood disorders: priming a target for new therapies. Pharmacol Ther. 2013; 138:18–37. https://doi.org/10.1016/j.pharmthera.2012.12.002 [PubMed]
- 18. de Mello Schier AR, de Oliveira Ribeiro NP, Coutinho DS, Machado S, Arias-Carrión O, Crippa JA, Zuardi AW, Nardi AE, Silva AC. Antidepressant-like and anxiolytic-like effects of cannabidiol: a chemical compound of Cannabis sativa. CNS Neurol Disord Drug Targets. 2014; 13:953–60. https://doi.org/10.2174/1871527313666140612114838 [PubMed]
- 19. Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002; 296:678–82. https://doi.org/10.1126/science.1063545 [PubMed]
- 20. Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012; 76:70–81. https://doi.org/10.1016/j.neuron.2012.09.020 [PubMed]
- 21. Silvestri C, Di Marzo V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 2013; 17:475–90. https://doi.org/10.1016/j.cmet.2013.03.001 [PubMed]
- 22. Atalay S, Jarocka-Karpowicz I, Skrzydlewska E. Antioxidative and Anti-Inflammatory Properties of Cannabidiol. Antioxidants (Basel). 2019; 9:21. https://doi.org/10.3390/antiox9010021 [PubMed]
- 23. Giordano S, Darley-Usmar V, Zhang J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2013; 2:82–90. https://doi.org/10.1016/j.redox.2013.12.013 [PubMed]
- 24. Franco-Iborra S, Vila M, Perier C. The Parkinson Disease Mitochondrial Hypothesis: Where Are We at? Neuroscientist. 2016; 22:266–77. https://doi.org/10.1177/1073858415574600 [PubMed]
- 25. Luque-Contreras D, Carvajal K, Toral-Rios D, Franco-Bocanegra D, Campos-Peña V. Oxidative stress and metabolic syndrome: cause or consequence of Alzheimer’s disease? Oxid Med Cell Longev. 2014; 2014:497802. https://doi.org/10.1155/2014/497802 [PubMed]
- 26. Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012; 15:349–57. https://doi.org/10.1038/nn.3028 [PubMed]
- 27. Sochocka M, Koutsouraki ES, Gasiorowski K, Leszek J. Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer’s disease: a new approach to therapy. CNS Neurol Disord Drug Targets. 2013; 12:870–81. https://doi.org/10.2174/18715273113129990072 [PubMed]
- 28. Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. 2017; 39:73–82. https://doi.org/10.1080/01616412.2016.1251711 [PubMed]
- 29. Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008; 7:97–109. https://doi.org/10.1016/S1474-4422(07)70327-7 [PubMed]
- 30. Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR. Oxidative stress and Parkinson’s disease. Front Neuroanat. 2015; 9:91. https://doi.org/10.3389/fnana.2015.00091 [PubMed]
- 31. Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993; 90:7915–22. https://doi.org/10.1073/pnas.90.17.7915 [PubMed]
- 32. Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993; 262:689–95. https://doi.org/10.1126/science.7901908 [PubMed]
- 33. Surace MJ, Block ML. Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell Mol Life Sci. 2012; 69:2409–27. https://doi.org/10.1007/s00018-012-1015-4 [PubMed]
- 34. Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis. 2013; 3:461–91. https://doi.org/10.3233/JPD-130230 [PubMed]
- 35. Puspita L, Chung SY, Shim JW. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain. 2017; 10:53. https://doi.org/10.1186/s13041-017-0340-9 [PubMed]
- 36. Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003 (Suppl 3); 53:S26–36. https://doi.org/10.1002/ana.10483 [PubMed]
- 37. Rahimmi A, Khosrobakhsh F, Izadpanah E, Moloudi MR, Hassanzadeh K. N-acetylcysteine prevents rotenone-induced Parkinson’s disease in rat: An investigation into the interaction of parkin and Drp1 proteins. Brain Res Bull. 2015; 113:34–40. https://doi.org/10.1016/j.brainresbull.2015.02.007 [PubMed]
- 38. Olanow CW, Schapira AH, LeWitt PA, Kieburtz K, Sauer D, Olivieri G, Pohlmann H, Hubble J. TCH346 as a neuroprotective drug in Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2006; 5:1013–20. https://doi.org/10.1016/S1474-4422(06)70602-0 [PubMed]
- 39. Lim KL, Tan JM. Role of the ubiquitin proteasome system in Parkinson’s disease. BMC Biochem. 2007 (Suppl 1); 8:S13. https://doi.org/10.1186/1471-2091-8-S1-S13 [PubMed]
- 40. Hassanzadeh K, Rahimmi A. Oxidative stress and neuroinflammation in the story of Parkinson’s disease: Could targeting these pathways write a good ending? J Cell Physiol. 2018; 234:23–32. https://doi.org/10.1002/jcp.26865 [PubMed]
- 41. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988; 38:1285–91. https://doi.org/10.1212/WNL.38.8.1285 [PubMed]
- 42. Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997; 12:25–31. [PubMed]
- 43. Gupta A, Dawson VL, Dawson TM. What causes cell death in Parkinson’s disease? Ann Neurol. 2008 (Suppl 2); 64:S3–15. https://doi.org/10.1002/ana.21573 [PubMed]
- 44. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010; 140:918–34. https://doi.org/10.1016/j.cell.2010.02.016 [PubMed]
- 45. Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013; 2013:480739. https://doi.org/10.1155/2013/480739 [PubMed]
- 46. Tufekci KU, Meuwissen R, Genc S, Genc K. Inflammation in Parkinson’s disease. Adv Protein Chem Struct Biol. 2012; 88:69–132. https://doi.org/10.1016/B978-0-12-398314-5.00004-0 [PubMed]
- 47. Rocha NP, de Miranda AS, Teixeira AL. Insights into Neuroinflammation in Parkinson’s Disease: From Biomarkers to Anti-Inflammatory Based Therapies. Biomed Res Int. 2015; 2015:628192. https://doi.org/10.1155/2015/628192 [PubMed]
- 48. Shih RH, Wang CY, Yang CM. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front Mol Neurosci. 2015; 8:77. https://doi.org/10.3389/fnmol.2015.00077 [PubMed]
- 49. Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, Ichinose H, Nagatsu T. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J Neural Transm (Vienna). 2000; 107:335–41. https://doi.org/10.1007/s007020050028 [PubMed]
- 50. Shimoji M, Pagan F, Healton EB, Mocchetti I. CXCR4 and CXCL12 expression is increased in the nigro-striatal system of Parkinson’s disease. Neurotox Res. 2009; 16:318–28. https://doi.org/10.1007/s12640-009-9076-3 [PubMed]
- 51. Yacoubian TA, Standaert DG. Targets for neuroprotection in Parkinson’s disease. Biochim Biophys Acta. 2009; 1792:676–87. https://doi.org/10.1016/j.bbadis.2008.09.009 [PubMed]
- 52. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010; 185:5577–85. https://doi.org/10.4049/jimmunol.1000548 [PubMed]
- 53. Russo I, Berti G, Plotegher N, Bernardo G, Filograna R, Bubacco L, Greggio E. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J Neuroinflammation. 2015; 12:230. https://doi.org/10.1186/s12974-015-0449-7 [PubMed]
- 54. López de Maturana R, Lang V, Zubiarrain A, Sousa A, Vázquez N, Gorostidi A, Águila J, López de Munain A, Rodríguez M, Sánchez-Pernaute R. Mutations in LRRK2 impair NF-κB pathway in iPSC-derived neurons. J Neuroinflammation. 2016; 13:295. https://doi.org/10.1186/s12974-016-0761-x [PubMed]
- 55. Moussaud S, Jones DR, Moussaud-Lamodière EL, Delenclos M, Ross OA, McLean PJ. Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener. 2014; 9:43. https://doi.org/10.1186/1750-1326-9-43 [PubMed]
- 56. Guerreiro PS, Gerhardt E, Lopes da Fonseca T, Bähr M, Outeiro TF, Eckermann K. LRRK2 Promotes Tau Accumulation, Aggregation and Release. Mol Neurobiol. 2016; 53:3124–35. https://doi.org/10.1007/s12035-015-9209-z [PubMed]
- 57. Russo I, Bubacco L, Greggio E. LRRK2 and neuroinflammation: partners in crime in Parkinson’s disease? J Neuroinflammation. 2014; 11:52. https://doi.org/10.1186/1742-2094-11-52 [PubMed]
- 58. Loh KM, van Amerongen R, Nusse R. Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev Cell. 2016; 38:643–55. https://doi.org/10.1016/j.devcel.2016.08.011 [PubMed]
- 59. Oren O, Smith BD. Eliminating Cancer Stem Cells by Targeting Embryonic Signaling Pathways. Stem Cell Rev Rep. 2017; 13:17–23. https://doi.org/10.1007/s12015-016-9691-3 [PubMed]
- 60. Al-Harthi L. Wnt/β-catenin and its diverse physiological cell signaling pathways in neurodegenerative and neuropsychiatric disorders. J Neuroimmune Pharmacol. 2012; 7:725–30. https://doi.org/10.1007/s11481-012-9412-x [PubMed]
- 61. Marchetti B, Pluchino S. Wnt your brain be inflamed? Yes, it Wnt!. Trends Mol Med. 2013; 19:144–56. https://doi.org/10.1016/j.molmed.2012.12.001 [PubMed]
- 62. Lecarpentier Y, Claes V, Duthoit G, Hébert JL. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front Physiol. 2014; 5:429. https://doi.org/10.3389/fphys.2014.00429 [PubMed]
- 63. Lecarpentier Y, Vallée A. Opposite Interplay between PPAR Gamma and Canonical Wnt/Beta-Catenin Pathway in Amyotrophic Lateral Sclerosis. Front Neurol. 2016; 7:100. https://doi.org/10.3389/fneur.2016.00100 [PubMed]
- 64. Vallée A, Lecarpentier Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front Neurosci. 2016; 10:459. https://doi.org/10.3389/fnins.2016.00459 [PubMed]
- 65. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998; 281:1509–12. https://doi.org/10.1126/science.281.5382.1509 [PubMed]
- 66. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999; 96:5522–27. https://doi.org/10.1073/pnas.96.10.5522 [PubMed]
- 67. Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009; 10:468–77. https://doi.org/10.1038/nrm2717 [PubMed]
- 68. Sharma C, Pradeep A, Wong L, Rana A, Rana B. Peroxisome proliferator-activated receptor gamma activation can regulate beta-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J Biol Chem. 2004; 279:35583–94. https://doi.org/10.1074/jbc.M403143200 [PubMed]
- 69. Rosi MC, Luccarini I, Grossi C, Fiorentini A, Spillantini MG, Prisco A, Scali C, Gianfriddo M, Caricasole A, Terstappen GC, Casamenti F. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J Neurochem. 2010; 112:1539–51. https://doi.org/10.1111/j.1471-4159.2009.06566.x [PubMed]
- 70. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012; 149:1192–205. https://doi.org/10.1016/j.cell.2012.05.012 [PubMed]
- 71. Inestrosa NC, Montecinos-Oliva C, Fuenzalida M. Wnt signaling: role in Alzheimer disease and schizophrenia. J Neuroimmune Pharmacol. 2012; 7:788–807. https://doi.org/10.1007/s11481-012-9417-5 [PubMed]
- 72. Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis. Oncotarget. 2017; 8:90579–604. https://doi.org/10.18632/oncotarget.21234 [PubMed]
- 73. Vallée A, Lecarpentier Y, Vallée JN. Hypothesis of Opposite Interplay Between the Canonical WNT/beta-catenin Pathway and PPAR Gamma in Primary Central Nervous System Lymphomas. Curr Issues Mol Biol. 2019; 31:1–20. https://doi.org/10.21775/cimb.031.001 [PubMed]
- 74. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997; 16:3797–804. https://doi.org/10.1093/emboj/16.13.3797 [PubMed]
- 75. Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci. 2010; 35:161–68. https://doi.org/10.1016/j.tibs.2009.10.002 [PubMed]
- 76. Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010; 11:539–51. https://doi.org/10.1038/nrn2870 [PubMed]
- 77. Ambacher KK, Pitzul KB, Karajgikar M, Hamilton A, Ferguson SS, Cregan SP. The JNK- and AKT/GSK3β- signaling pathways converge to regulate Puma induction and neuronal apoptosis induced by trophic factor deprivation. PLoS One. 2012; 7:e46885. https://doi.org/10.1371/journal.pone.0046885 [PubMed]
- 78. Orellana AM, Vasconcelos AR, Leite JA, de Sá Lima L, Andreotti DZ, Munhoz CD, Kawamoto EM, Scavone C. Age-related neuroinflammation and changes in AKT-GSK-3β and WNT/ β-CATENIN signaling in rat hippocampus. Aging (Albany NY). 2015; 7:1094–111. https://doi.org/10.18632/aging.100853 [PubMed]
- 79. Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci. 2010; 11:77–86. https://doi.org/10.1038/nrn2755 [PubMed]
- 80. Berwick DC, Harvey K. LRRK2 signaling pathways: the key to unlocking neurodegeneration? Trends Cell Biol. 2011; 21:257–65. https://doi.org/10.1016/j.tcb.2011.01.001 [PubMed]
- 81. L’episcopo F, Serapide MF, Tirolo C, Testa N, Caniglia S, Morale MC, Pluchino S, Marchetti B. A Wnt1 regulated Frizzled-1/β-Catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: Therapeutical relevance for neuron survival and neuroprotection. Mol Neurodegener. 2011; 6:49. https://doi.org/10.1186/1750-1326-6-49 [PubMed]
- 82. Häbig K, Walter M, Poths S, Riess O, Bonin M. RNA interference of LRRK2-microarray expression analysis of a Parkinson’s disease key player. Neurogenetics. 2008; 9:83–94. https://doi.org/10.1007/s10048-007-0114-0 [PubMed]
- 83. Libro R, Bramanti P, Mazzon E. The role of the Wnt canonical signaling in neurodegenerative diseases. Life Sci. 2016; 158:78–88. https://doi.org/10.1016/j.lfs.2016.06.024 [PubMed]
- 84. Zhou T, Zu G, Zhang X, Wang X, Li S, Gong X, Liang Z, Zhao J. Neuroprotective effects of ginsenoside Rg1 through the Wnt/β-catenin signaling pathway in both in vivo and in vitro models of Parkinson’s disease. Neuropharmacology. 2016; 101:480–89. https://doi.org/10.1016/j.neuropharm.2015.10.024 [PubMed]
- 85. L’Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Deleidi M, Serapide MF, Pluchino S, Marchetti B. Plasticity of subventricular zone neuroprogenitors in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of Parkinson’s disease involves cross talk between inflammatory and Wnt/β-catenin signaling pathways: functional consequences for neuroprotection and repair. J Neurosci. 2012; 32:2062–85. https://doi.org/10.1523/JNEUROSCI.5259-11.2012 [PubMed]
- 86. Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab. 2005; 16:183–89. https://doi.org/10.1016/j.tem.2005.03.010 [PubMed]
- 87. Almeida M, Ambrogini E, Han L, Manolagas SC, Jilka RL. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-gamma expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J Biol Chem. 2009; 284:27438–48. https://doi.org/10.1074/jbc.M109.023572 [PubMed]
- 88. Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005; 308:1181–84. https://doi.org/10.1126/science.1109083 [PubMed]
- 89. Hoogeboom D, Essers MA, Polderman PE, Voets E, Smits LM, Burgering BM. Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J Biol Chem. 2008; 283:9224–30. https://doi.org/10.1074/jbc.M706638200 [PubMed]
- 90. Reif K, Burgering BM, Cantrell DA. Phosphatidylinositol 3-kinase links the interleukin-2 receptor to protein kinase B and p70 S6 kinase. J Biol Chem. 1997; 272:14426–33. https://doi.org/10.1074/jbc.272.22.14426 [PubMed]
- 91. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999; 96:857–68. https://doi.org/10.1016/S0092-8674(00)80595-4 [PubMed]
- 92. Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002; 168:5024–31. https://doi.org/10.4049/jimmunol.168.10.5024 [PubMed]
- 93. Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002; 22:7842–52. https://doi.org/10.1128/MCB.22.22.7842-7852.2002 [PubMed]
- 94. Fernández de Mattos S, Essafi A, Soeiro I, Pietersen AM, Birkenkamp KU, Edwards CS, Martino A, Nelson BH, Francis JM, Jones MC, Brosens JJ, Coffer PJ, Lam EW. FoxO3a and BCR-ABL regulate cyclin D2 transcription through a STAT5/BCL6-dependent mechanism. Mol Cell Biol. 2004; 24:10058–71. https://doi.org/10.1128/MCB.24.22.10058-10071.2004 [PubMed]
- 95. Manolopoulos KN, Klotz LO, Korsten P, Bornstein SR, Barthel A. Linking Alzheimer’s disease to insulin resistance: the FoxO response to oxidative stress. Mol Psychiatry. 2010; 15:1046–52. https://doi.org/10.1038/mp.2010.17 [PubMed]
- 96. Shang YC, Chong ZZ, Hou J, Maiese K. Wnt1, FoxO3a, and NF-kappaB oversee microglial integrity and activation during oxidant stress. Cell Signal. 2010; 22:1317–29. https://doi.org/10.1016/j.cellsig.2010.04.009 [PubMed]
- 97. Halleskog C, Mulder J, Dahlström J, Mackie K, Hortobágyi T, Tanila H, Kumar Puli L, Färber K, Harkany T, Schulte G. WNT signaling in activated microglia is proinflammatory. Glia. 2011; 59:119–31. https://doi.org/10.1002/glia.21081 [PubMed]
- 98. Ma B, Hottiger MO. Crosstalk between Wnt/β-Catenin and NF-κB Signaling Pathway during Inflammation. Front Immunol. 2016; 7:378. https://doi.org/10.3389/fimmu.2016.00378 [PubMed]
- 99. Mitchell S, Vargas J, Hoffmann A. Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med. 2016; 8:227–41. https://doi.org/10.1002/wsbm.1331 [PubMed]
- 100. Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, Li Y, Lin SY, Hung MC. beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell. 2002; 2:323–34. https://doi.org/10.1016/S1535-6108(02)00154-X [PubMed]
- 101. Liu J, Liao Y, Ma K, Wang Y, Zhang G, Yang R, Deng J. PI3K is required for the physical interaction and functional inhibition of NF-κB by β-catenin in colorectal cancer cells. Biochem Biophys Res Commun. 2013; 434:760–66. https://doi.org/10.1016/j.bbrc.2013.03.135 [PubMed]
- 102. Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005; 6:777–84. https://doi.org/10.1038/ni1221 [PubMed]
- 103. Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, Pulendran B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. 2010; 329:849–53. https://doi.org/10.1126/science.1188510 [PubMed]
- 104. Cho HH, Song JS, Yu JM, Yu SS, Choi SJ, Kim DH, Jung JS. Differential effect of NF-kappaB activity on beta-catenin/Tcf pathway in various cancer cells. FEBS Lett. 2008; 582:616–22. https://doi.org/10.1016/j.febslet.2008.01.029 [PubMed]
- 105. Fliniaux I, Mikkola ML, Lefebvre S, Thesleff I. Identification of dkk4 as a target of Eda-A1/Edar pathway reveals an unexpected role of ectodysplasin as inhibitor of Wnt signalling in ectodermal placodes. Dev Biol. 2008; 320:60–71. https://doi.org/10.1016/j.ydbio.2008.04.023 [PubMed]
- 106. Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000; 406:86–90. https://doi.org/10.1038/35017574 [PubMed]
- 107. Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010; 31:24–31. https://doi.org/10.1016/j.it.2009.09.007 [PubMed]
- 108. Russo E, Guy GW. A tale of two cannabinoids: the therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med Hypotheses. 2006; 66:234–46. https://doi.org/10.1016/j.mehy.2005.08.026 [PubMed]
- 109. Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimarães FS. Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders. Philos Trans R Soc Lond B Biol Sci. 2012; 367:3364–78. https://doi.org/10.1098/rstb.2011.0389 [PubMed]
- 110. Pertwee RG. Endocannabinoids and Their Pharmacological Actions. Handb Exp Pharmacol. 2015; 231:1–37. https://doi.org/10.1007/978-3-319-20825-1_1 [PubMed]
- 111. Bergamaschi MM, Queiroz RH, Zuardi AW, Crippa JA. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr Drug Saf. 2011; 6:237–49. https://doi.org/10.2174/157488611798280924 [PubMed]
- 112. Iffland K, Grotenhermen F. An Update on Safety and Side Effects of Cannabidiol: A Review of Clinical Data and Relevant Animal Studies. Cannabis Cannabinoid Res. 2017; 2:139–54. https://doi.org/10.1089/can.2016.0034 [PubMed]
- 113. Devinsky O, Cilio MR, Cross H, Fernandez-Ruiz J, French J, Hill C, Katz R, Di Marzo V, Jutras-Aswad D, Notcutt WG, Martinez-Orgado J, Robson PJ, Rohrback BG, et al. Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia. 2014; 55:791–802. https://doi.org/10.1111/epi.12631 [PubMed]
- 114. Emamian ES. AKT/GSK3 signaling pathway and schizophrenia. Front Mol Neurosci. 2012; 5:33. https://doi.org/10.3389/fnmol.2012.00033 [PubMed]
- 115. Renard J, Norris C, Rushlow W, Laviolette SR. Neuronal and molecular effects of cannabidiol on the mesolimbic dopamine system: Implications for novel schizophrenia treatments. Neurosci Biobehav Rev. 2017; 75:157–65. https://doi.org/10.1016/j.neubiorev.2017.02.006 [PubMed]
- 116. Cassano T, Serviddio G, Gaetani S, Romano A, Dipasquale P, Cianci S, Bellanti F, Laconca L, Romano AD, Padalino I, LaFerla FM, Nicoletti F, Cuomo V, Vendemiale G. Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol Aging. 2012; 33:1121.e1–12. https://doi.org/10.1016/j.neurobiolaging.2011.09.021 [PubMed]
- 117. Giudetti AM, Salzet M, Cassano T. Oxidative Stress in Aging Brain: Nutritional and Pharmacological Interventions for Neurodegenerative Disorders. Oxid Med Cell Longev. 2018; 2018:3416028. https://doi.org/10.1155/2018/3416028 [PubMed]
- 118. Izzo AA, Borrelli F, Capasso R, Di Marzo V, Mechoulam R. Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol Sci. 2009; 30:515–27. https://doi.org/10.1016/j.tips.2009.07.006 [PubMed]
- 119. Costa B, Colleoni M, Conti S, Parolaro D, Franke C, Trovato AE, Giagnoni G. Oral anti-inflammatory activity of cannabidiol, a non-psychoactive constituent of cannabis, in acute carrageenan-induced inflammation in the rat paw. Naunyn Schmiedebergs Arch Pharmacol. 2004; 369:294–99. https://doi.org/10.1007/s00210-004-0871-3 [PubMed]
- 120. Lus G, Cantello R, Danni MC, Rini A, Sarchielli P, Tassinari T, Signoriello E. Palatability and oral cavity tolerability of THC:CBD oromucosal spray and possible improvement measures in multiple sclerosis patients with resistant spasticity: a pilot study. Neurodegener Dis Manag. 2018; 8:105–13. https://doi.org/10.2217/nmt-2017-0056 [PubMed]
- 121. Cortesi M, Fusar-Poli P. Potential therapeutical effects of cannabidiol in children with pharmacoresistant epilepsy. Med Hypotheses. 2007; 68:920–21. https://doi.org/10.1016/j.mehy.2006.09.030 [PubMed]
- 122. Murillo-Rodríguez E, Millán-Aldaco D, Palomero-Rivero M, Mechoulam R, Drucker-Colín R. Cannabidiol, a constituent of Cannabis sativa, modulates sleep in rats. FEBS Lett. 2006; 580:4337–45. https://doi.org/10.1016/j.febslet.2006.04.102 [PubMed]
- 123. Parker LA, Rock EM, Limebeer CL. Regulation of nausea and vomiting by cannabinoids. Br J Pharmacol. 2011; 163:1411–22. https://doi.org/10.1111/j.1476-5381.2010.01176.x [PubMed]
- 124. Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, Klosterkötter J, Hellmich M, Koethe D. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. 2012; 2:e94. https://doi.org/10.1038/tp.2012.15 [PubMed]
- 125. Gomes FV, Resstel LB, Guimarães FS. The anxiolytic-like effects of cannabidiol injected into the bed nucleus of the stria terminalis are mediated by 5-HT1A receptors. Psychopharmacology (Berl). 2011; 213:465–73. https://doi.org/10.1007/s00213-010-2036-z [PubMed]
- 126. Zanelati TV, Biojone C, Moreira FA, Guimarães FS, Joca SR. Antidepressant-like effects of cannabidiol in mice: possible involvement of 5-HT1A receptors. Br J Pharmacol. 2010; 159:122–28. https://doi.org/10.1111/j.1476-5381.2009.00521.x [PubMed]
- 127. Lastres-Becker I, Molina-Holgado F, Ramos JA, Mechoulam R, Fernández-Ruiz J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson’s disease. Neurobiol Dis. 2005; 19:96–107. https://doi.org/10.1016/j.nbd.2004.11.009 [PubMed]
- 128. García C, Palomo-Garo C, García-Arencibia M, Ramos J, Pertwee R, Fernández-Ruiz J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ9-THCV in animal models of Parkinson’s disease. Br J Pharmacol. 2011; 163:1495–506. https://doi.org/10.1111/j.1476-5381.2011.01278.x [PubMed]
- 129. Balash Y, Bar-Lev Schleider L, Korczyn AD, Shabtai H, Knaani J, Rosenberg A, Baruch Y, Djaldetti R, Giladi N, Gurevich T. Medical Cannabis in Parkinson Disease: Real-Life Patients’ Experience. Clin Neuropharmacol. 2017; 40:268–72. https://doi.org/10.1097/WNF.0000000000000246 [PubMed]
- 130. Lotan I, Treves TA, Roditi Y, Djaldetti R. Cannabis (medical marijuana) treatment for motor and non-motor symptoms of Parkinson disease: an open-label observational study. Clin Neuropharmacol. 2014; 37:41–44. https://doi.org/10.1097/WNF.0000000000000016 [PubMed]
- 131. Casarejos MJ, Perucho J, Gomez A, Muñoz MP, Fernandez-Estevez M, Sagredo O, Fernandez Ruiz J, Guzman M, de Yebenes JG, Mena MA. Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. J Alzheimers Dis. 2013; 35:525–39. https://doi.org/10.3233/JAD-130050 [PubMed]
- 132. Crippa JA, Hallak JE, Zuardi AW, Guimarães FS, Tumas V, Dos Santos RG. Is cannabidiol the ideal drug to treat non-motor Parkinson’s disease symptoms? Eur Arch Psychiatry Clin Neurosci. 2019; 269:121–33. https://doi.org/10.1007/s00406-019-00982-6 [PubMed]
- 133. Martinez AA, Morgese MG, Pisanu A, Macheda T, Paquette MA, Seillier A, Cassano T, Carta AR, Giuffrida A. Activation of PPAR gamma receptors reduces levodopa-induced dyskinesias in 6-OHDA-lesioned rats. Neurobiol Dis. 2015; 74:295–304. https://doi.org/10.1016/j.nbd.2014.11.024 [PubMed]
- 134. García-Arencibia M, González S, de Lago E, Ramos JA, Mechoulam R, Fernández-Ruiz J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007; 1134:162–70. https://doi.org/10.1016/j.brainres.2006.11.063 [PubMed]
- 135. Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA. 1998; 95:8268–73. https://doi.org/10.1073/pnas.95.14.8268 [PubMed]
- 136. Thompson SL, Dulawa SC. Dissecting the roles of β-arrestin2 and GSK-3 signaling in 5-HT1BR-mediated perseverative behavior and prepulse inhibition deficits in mice. PLoS One. 2019; 14:e0211239. https://doi.org/10.1371/journal.pone.0211239 [PubMed]
- 137. Giese KP. GSK-3: a key player in neurodegeneration and memory. IUBMB Life. 2009; 61:516–21. https://doi.org/10.1002/iub.187 [PubMed]
- 138. Vallée A, Vallée JN, Lecarpentier Y. PPARγ agonists: potential treatment for autism spectrum disorder by inhibiting the canonical WNT/β-catenin pathway. Mol Psychiatry. 2019; 24:643–52. https://doi.org/10.1038/s41380-018-0131-4 [PubMed]
- 139. Vallée A, Lecarpentier Y, Vallée JN. Targeting the Canonical WNT/β-Catenin Pathway in Cancer Treatment Using Non-Steroidal Anti-Inflammatory Drugs. Cells. 2019; 8:726. https://doi.org/10.3390/cells8070726 [PubMed]
- 140. Vallée A, Vallée JN. Warburg effect hypothesis in autism Spectrum disorders. Mol Brain. 2018; 11:1. https://doi.org/10.1186/s13041-017-0343-6 [PubMed]
- 141. Libro R, Diomede F, Scionti D, Piattelli A, Grassi G, Pollastro F, Bramanti P, Mazzon E, Trubiani O. Cannabidiol Modulates the Expression of Alzheimer’s Disease-Related Genes in Mesenchymal Stem Cells. Int J Mol Sci. 2016; 18:26. https://doi.org/10.3390/ijms18010026 [PubMed]
- 142. Giacoppo S, Pollastro F, Grassi G, Bramanti P, Mazzon E. Target regulation of PI3K/Akt/mTOR pathway by cannabidiol in treatment of experimental multiple sclerosis. Fitoterapia. 2017; 116:77–84. https://doi.org/10.1016/j.fitote.2016.11.010 [PubMed]
- 143. Hernández F, Gómez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J. GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol. 2010; 223:322–25. https://doi.org/10.1016/j.expneurol.2009.09.011 [PubMed]
- 144. Ozaita A, Puighermanal E, Maldonado R. Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J Neurochem. 2007; 102:1105–14. https://doi.org/10.1111/j.1471-4159.2007.04642.x [PubMed]
- 145. Trazzi S, Steger M, Mitrugno VM, Bartesaghi R, Ciani E. CB1 cannabinoid receptors increase neuronal precursor proliferation through AKT/glycogen synthase kinase-3beta/beta-catenin signaling. J Biol Chem. 2010; 285:10098–109. https://doi.org/10.1074/jbc.M109.043711 [PubMed]
- 146. Yue X, Lan F, Yang W, Yang Y, Han L, Zhang A, Liu J, Zeng H, Jiang T, Pu P, Kang C. Interruption of β-catenin suppresses the EGFR pathway by blocking multiple oncogenic targets in human glioma cells. Brain Res. 2010; 1366:27–37. https://doi.org/10.1016/j.brainres.2010.10.032 [PubMed]
- 147. Gómez Del Pulgar T, De Ceballos ML, Guzmán M, Velasco G. Cannabinoids protect astrocytes from ceramide-induced apoptosis through the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2002; 277:36527–33. https://doi.org/10.1074/jbc.M205797200 [PubMed]
- 148. Molina-Holgado E, Vela JM, Arévalo-Martín A, Almazán G, Molina-Holgado F, Borrell J, Guaza C. Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci. 2002; 22:9742–53. https://doi.org/10.1523/JNEUROSCI.22-22-09742.2002 [PubMed]
- 149. Stone NL, Murphy AJ, England TJ, O’Sullivan SE. A systematic review of minor phytocannabinoids with promising neuroprotective potential. Br J Pharmacol. 2020; 177:4330–52. https://doi.org/10.1111/bph.15185 [PubMed]
- 150. Borges RS, Batista J
Jr , Viana RB, Baetas AC, Orestes E, Andrade MA, Honório KM, da Silva AB. Understanding the molecular aspects of tetrahydrocannabinol and cannabidiol as antioxidants. Molecules. 2013; 18:12663–74. https://doi.org/10.3390/molecules181012663 [PubMed] - 151. Rajesh M, Mukhopadhyay P, Bátkai S, Haskó G, Liaudet L, Drel VR, Obrosova IG, Pacher P. Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am J Physiol Heart Circ Physiol. 2007; 293:H610–19. https://doi.org/10.1152/ajpheart.00236.2007 [PubMed]
- 152. Pan H, Mukhopadhyay P, Rajesh M, Patel V, Mukhopadhyay B, Gao B, Haskó G, Pacher P. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J Pharmacol Exp Ther. 2009; 328:708–14. https://doi.org/10.1124/jpet.108.147181 [PubMed]
- 153. Fouad AA, Albuali WH, Al-Mulhim AS, Jresat I. Cardioprotective effect of cannabidiol in rats exposed to doxorubicin toxicity. Environ Toxicol Pharmacol. 2013; 36:347–57. https://doi.org/10.1016/j.etap.2013.04.018 [PubMed]
- 154. Hamelink C, Hampson A, Wink DA, Eiden LE, Eskay RL. Comparison of cannabidiol, antioxidants, and diuretics in reversing binge ethanol-induced neurotoxicity. J Pharmacol Exp Ther. 2005; 314:780–88. https://doi.org/10.1124/jpet.105.085779 [PubMed]
- 155. da Silva VK, de Freitas BS, Garcia RC, Monteiro RT, Hallak JE, Zuardi AW, Crippa JA, Schröder N. Antiapoptotic effects of cannabidiol in an experimental model of cognitive decline induced by brain iron overload. Transl Psychiatry. 2018; 8:176. https://doi.org/10.1038/s41398-018-0232-5 [PubMed]
- 156. Vomund S, Schäfer A, Parnham MJ, Brüne B, von Knethen A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int J Mol Sci. 2017; 18:2772. https://doi.org/10.3390/ijms18122772 [PubMed]
- 157. Rajesh M, Mukhopadhyay P, Bátkai S, Patel V, Saito K, Matsumoto S, Kashiwaya Y, Horváth B, Mukhopadhyay B, Becker L, Haskó G, Liaudet L, Wink DA, et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol. 2010; 56:2115–25. https://doi.org/10.1016/j.jacc.2010.07.033 [PubMed]
- 158. Costa B, Trovato AE, Comelli F, Giagnoni G, Colleoni M. The non-psychoactive cannabis constituent cannabidiol is an orally effective therapeutic agent in rat chronic inflammatory and neuropathic pain. Eur J Pharmacol. 2007; 556:75–83. https://doi.org/10.1016/j.ejphar.2006.11.006 [PubMed]
- 159. Wu HY, Jan TR. Cannabidiol hydroxyquinone-induced apoptosis of splenocytes is mediated predominantly by thiol depletion. Toxicol Lett. 2010; 195:68–74. https://doi.org/10.1016/j.toxlet.2010.02.012 [PubMed]
- 160. Gęgotek A, Ambrożewicz E, Jastrząb A, Jarocka-Karpowicz I, Skrzydlewska E. Rutin and ascorbic acid cooperation in antioxidant and antiapoptotic effect on human skin keratinocytes and fibroblasts exposed to UVA and UVB radiation. Arch Dermatol Res. 2019; 311:203–19. https://doi.org/10.1007/s00403-019-01898-w [PubMed]
- 161. Pertwee RG. The pharmacology of cannabinoid receptors and their ligands: an overview. Int J Obes (Lond). 2006 (Suppl 1); 30:S13–18. https://doi.org/10.1038/sj.ijo.0803272 [PubMed]
- 162. Wang S, Xu Q, Shu G, Wang L, Gao P, Xi Q, Zhang Y, Jiang Q, Zhu X. N-Oleoyl glycine, a lipoamino acid, stimulates adipogenesis associated with activation of CB1 receptor and Akt signaling pathway in 3T3-L1 adipocyte. Biochem Biophys Res Commun. 2015; 466:438–43. https://doi.org/10.1016/j.bbrc.2015.09.046 [PubMed]
- 163. Wang Y, Mukhopadhyay P, Cao Z, Wang H, Feng D, Haskó G, Mechoulam R, Gao B, Pacher P. Cannabidiol attenuates alcohol-induced liver steatosis, metabolic dysregulation, inflammation and neutrophil-mediated injury. Sci Rep. 2017; 7:12064. https://doi.org/10.1038/s41598-017-10924-8 [PubMed]
- 164. Hou Y, Moreau F, Chadee K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat Commun. 2012; 3:1300. https://doi.org/10.1038/ncomms2270 [PubMed]
- 165. Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003; 144:2201–07. https://doi.org/10.1210/en.2003-0288 [PubMed]
- 166. Marx N, Duez H, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors and atherogenesis: regulators of gene expression in vascular cells. Circ Res. 2004; 94:1168–78. https://doi.org/10.1161/01.RES.0000127122.22685.0A [PubMed]
- 167. Cunard R, Ricote M, DiCampli D, Archer DC, Kahn DA, Glass CK, Kelly CJ. Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. J Immunol. 2002; 168:2795–802. https://doi.org/10.4049/jimmunol.168.6.2795 [PubMed]
- 168. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998; 391:79–82. https://doi.org/10.1038/34178 [PubMed]
- 169. Giannini S, Serio M, Galli A. Pleiotropic effects of thiazolidinediones: taking a look beyond antidiabetic activity. J Endocrinol Invest. 2004; 27:982–91. https://doi.org/10.1007/BF03347546 [PubMed]
- 170. Vallée A, Lecarpentier Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front Immunol. 2018; 9:745. https://doi.org/10.3389/fimmu.2018.00745 [PubMed]
- 171. Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Thermodynamics in Gliomas: Interactions between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma. Front Physiol. 2017; 8:352. https://doi.org/10.3389/fphys.2017.00352 [PubMed]
- 172. Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Demyelination in Multiple Sclerosis: Reprogramming Energy Metabolism and Potential PPARγ Agonist Treatment Approaches. Int J Mol Sci. 2018; 19:1212. https://doi.org/10.3390/ijms19041212 [PubMed]
- 173. Park KS, Lee RD, Kang SK, Han SY, Park KL, Yang KH, Song YS, Park HJ, Lee YM, Yun YP, Oh KW, Kim DJ, Yun YW, et al. Neuronal differentiation of embryonic midbrain cells by upregulation of peroxisome proliferator-activated receptor-gamma via the JNK-dependent pathway. Exp Cell Res. 2004; 297:424–33. https://doi.org/10.1016/j.yexcr.2004.03.034 [PubMed]
- 174. Vallée A, Lecarpentier Y, Vallée JN. Thermodynamic Aspects and Reprogramming Cellular Energy Metabolism during the Fibrosis Process. Int J Mol Sci. 2017; 18:2537. https://doi.org/10.3390/ijms18122537 [PubMed]
- 175. Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Reprogramming energetic metabolism in Alzheimer’s disease. Life Sci. 2018; 193:141–52. https://doi.org/10.1016/j.lfs.2017.10.033 [PubMed]
- 176. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001; 65:391–426. https://doi.org/10.1016/S0301-0082(01)00011-9 [PubMed]
- 177. Jeon M, Rahman N, Kim YS. Wnt/β-catenin signaling plays a distinct role in methyl gallate-mediated inhibition of adipogenesis. Biochem Biophys Res Commun. 2016; 479:22–27. https://doi.org/10.1016/j.bbrc.2016.08.178 [PubMed]
- 178. Gustafson B, Eliasson B, Smith U. Thiazolidinediones increase the wingless-type MMTV integration site family (WNT) inhibitor Dickkopf-1 in adipocytes: a link with osteogenesis. Diabetologia. 2010; 53:536–40. https://doi.org/10.1007/s00125-009-1615-1 [PubMed]