




Introduction
The third most common type of gastrointestinal cancer is colorectal cancer (CRC). It is also the third leading cause of cancer-induced deaths worldwide. It affects more than 900,000 patients each year [1–3]. The more progressive screening techniques and treatment modalities were used to decline the mortality rate of CRC. The metastatic colorectal cancer (mCRC) patients are used with various combinations of chemotherapeutic drugs and biologics, including vascular endothelial growth factors inhibitors (VEGF inhibitors; e.g., bevacizumab) and epidermal growth factor receptors inhibitors (EGFR inhibitors; e.g., cetuximab) [4]. The current standard treatment improves outcomes in most patients with mCRC but fails to provide prominent benefits in a notable proportion of individuals who experience drug-associated toxicities. Thus, it is imperative to develop predictive biomarkers that can help select patients who might benefit from such treatment. Accordingly, patients who might not obtain any benefit from such treatment can avoid drug-related toxicity and receive alternative treatment [5].
Angiogenesis is a complex process and a critical pace in the progression of malignancies. One of the symbols of cancer progression is angiogenesis [6, 7]. Apoptosis is a critical homeostatic mechanism that helps keep cell populations during normal cell development. Disruption in apoptosis regulation might lead to cancer [8]. MicroRNAs (miRNAs), noncoding RNAs, mediate the genetic expressions at the posttranscriptional level and play essential roles in tumorigenesis, cancer progression, and clinical therapy in various malignancies, one of which is CRC [2, 9, 10]. In addition, miRNAs, such as oncogenes and tumor suppressor genes, contribute to the proliferation, apoptosis, angiogenesis, invasion, and tumor metastasis of various cancers [11–13]. Hypoxia has been implicated in the pathogenesis of CRC and other malignancies. It can prevent apoptosis and result in cancer growth, anti-apoptosis, recurrence, and poor survival [14]. Li and his colleagues suggested in a review that miR-148a inhibits the proliferation, apoptosis, metastasis, and invasion of cancer cells by directly targeting ROCK1 and BCL-2 [15, 16]. A study demonstrated that miR-148a suppresses the epithelial-mesenchymal transition (EMT) of hepatocellular carcinoma by targeting c-Met [17]. We have previously observed that miR-148a inhibits tumorigenesis and reduces the likelihood of early CRC recurrence [18], improves the response to chemoradiation and increases apoptosis by directly targeting c-Met in patients with rectal cancer [19], indirectly inhibits VEGF secretion by targeting HIF-1α [7], and exhibits the apoptotic effect of alterations of myeloid cell leukemia 1 (Mcl-1) expression on CRC [20].
Xu et al. demonstrated in 2018 that chemokine CC ligand 19 (CCL19) inhibits CRC angiogenesis by promoting miR-206 and thereby inhibiting the Met/ERK/HIF-1α/VEGF-A pathway [21]. Gluck et al. proved that Met regulates HIF-1α levels via a protein translation mechanism [22]. A previous study found that the Rho/ROCK pathway is essential to HIF-1α expression in ovarian cancer cell lines and is an upstream regulator of HIF-1α accumulation in ovarian cancer [23]. In 2020, Wu et al. reported that HIF-1α could promote Mcl-1 expression to act as a transcription factor by directly target the promoter region of Mcl-1 [24].
In this current study, we supposed that miR-148a directly targets ROCK1 and c-Met to decrease angiogenesis and increase the apoptosis of colon cancer cells by inhibiting the secretion of VEGF and Mcl-1 through downregulation of HIF-1α under hypoxia. We also searched out the potential synergistic effects of the combination of miR-148a and bevacizumab.
Results
miR-148a posttranscriptional directly reduced ROCK1 and c-Met expression by targeting its 3′-UTR
We explored the mechanisms underlying the antimetastatic functions of miR-148a. The predicted target genes of miR-148a were gained from the TargetScan database (https://targetscan.org) and the isomiRTar portal (https://isomirtar.hse.ru/), especially those that can suppress angiogenesis and apoptosis in cancer cells. Thus, two candidate genes, ROCK1 and c-Met, were selected (Supplementary Figure 1A, 1B).
ROCK1, an effector kinase of Rho GTPases, plays a vital role in the regulation of cancer invasion and metastasis [25, 26]. Two possible binding sites on the 3′-UTR sequence of ROCK1 for miR-148a were identified using TargetScan (https://targetscan.org). A series of 3′-UTR fragments of ROCK1 including the full sequence, binding site1, binding site2, and their corresponding mutant counterparts were directly fused downstream of the firefly luciferase gene to assess whether ROCK1 is a direct target of miR-148a. We co-transfected the mutant luciferase 3′-UTR construct into HCT116 and HCT116-miR-148a cells (Figure 1A). The luciferase activity of ROCK1 was significantly decreased in the HCT116-miR-148a cell line transfected with wild-type ROCK1 3′-UTR (P = 0.001) but not in that transfected with mutant ROCK1 3′-UTR (P = 0.09; Figure 1B). A comparison of the mRNA expression of ROCK1 between the HCT116 cells and HCT116-miR-148a cells revealed that miR-148a remarkably downregulated the expression of ROCK1 mRNA in the HCT116-miR-148a cells (P = 0.001; Figure 1C).
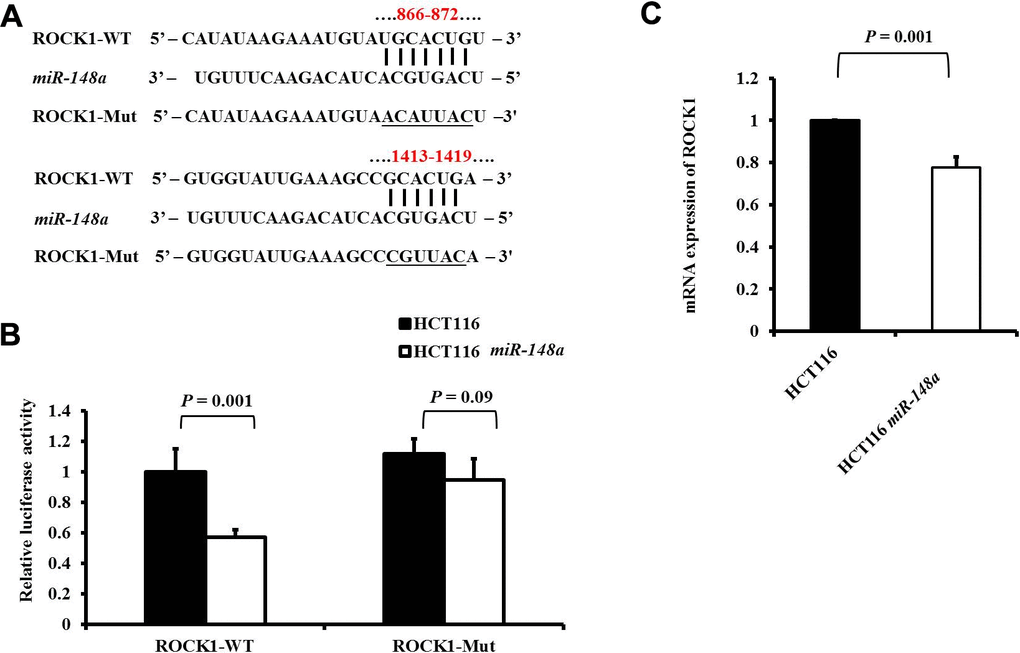
Figure 1. ROCK1 is a direct target of miR-148a in HCT-116 cells. (A) Two putative miR-148a-binding sites in ROCK1 3′-UTR and the two corresponding mutant binding sites (underlined) are shown; (B) miR-148a overexpression suppressed the activity of firefly luciferase that carried the wild-type (P = 0.001) but not mutant 3′-UTR of ROCK1; (C) The mRNA levels of ROCK1 were determined using qRT-PCR in stable HCT116 and HCT116-miR-148a cell lines, and the difference was significant in the HCT116-miR-148a cell line (P = 0.001).
Met, a tyrosine kinase receptor for hepatocyte growth factor (HGF), plays an important role in HGF/Met/Snail signaling in the EMT and metastasis [27, 28]. The wild-type or mutant 3′-UTR fragments of Met were cloned downstream of the firefly luciferase gene to determine whether Met is a direct target of miR-148a (Figure 2A). It demonstrated by a luciferase reporter assay that the overexpression of miR-148a in the HT29 cell line significantly attenuated firefly luciferase activity relative to that in the cell line transfected with the wild-type 3′-UTR of Met (P < 0.001; Figure 2B), when the predicted 3′-UTR-binding site was mutated and the effect was abolished (P = 0.11; Figure 2B). A comparison of the mRNA expression of c-Met between the HT29 cells and HT29-miR-148a cells revealed that miR-148a remarkably downregulated the expression of c-Met mRNA (P < 0.001; Figure 2C) in the HT29-miR-148a cells.
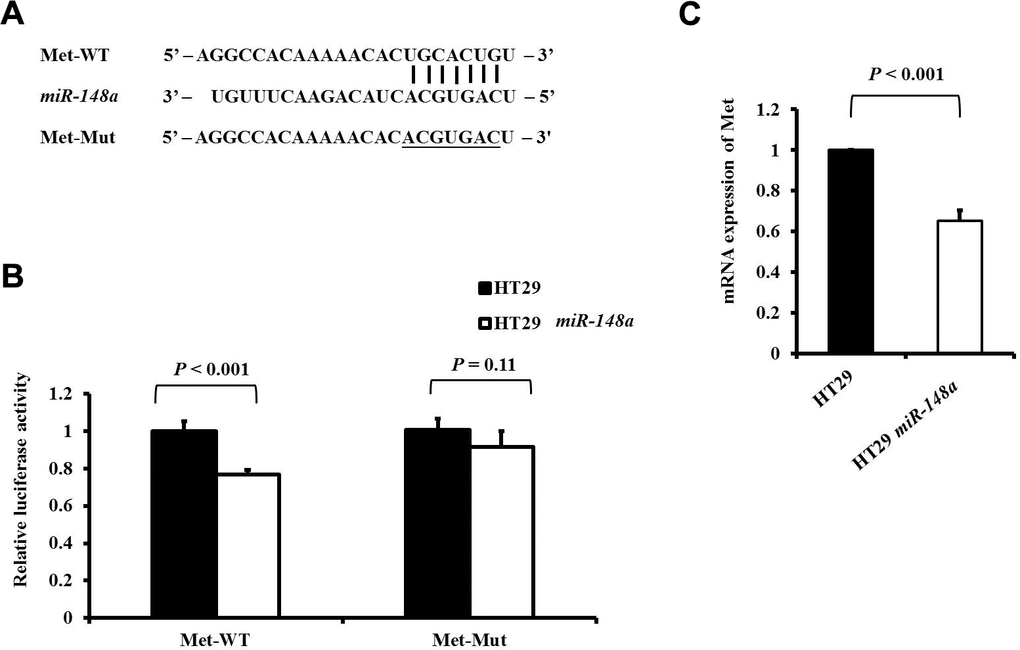
Figure 2. Met is a direct target of miR-148a in HT-29 cells. (A) miR-148a and its putative binding sequences in the 3′-UTR of Met. Mutations were generated at the complementary site (underlined) that binds to the seed region of miR-148a; (B) miR-148a overexpression suppressed the activity of firefly luciferase that carried the wild-type (P < 0.001) but not mutant 3′-UTR of Met; (C) The mRNA levels of Met were determined using qRT-PCR in stable HT29 and HT29-miR-148a cell lines, and the difference was significant in the HT29-miR-148a cell line (P < 0.001).
miR-148a directly targets ROCK1 in the HCT116 cell line to curb the expression of HIF-1α and Mcl-1 under hypoxia
To examine the functions of miR-148a on the activation of ROCK1, HIF-1α, and Mcl-1 under hypoxia, we analyzed the protein expression of ROCK1, HIF-1α, and Mcl-1 in the HCT116, HCT116 + bevacizumab, HCT116-miR-148a, and HCT116-miR-148a + bevacizumab cell lines, separately. The protein expression of ROCK1 was significantly decreased in the HCT116-miR-148a cells compared with that in the HCT116 cells (Figure 3A, 3B, P = 0.002), but not in the HCT116 + bevacizumab cells (P = 0.58, Figure 3A, 3B). Similarly, the protein expression of HIF-1α and Mcl-1 (all P < 0.001; Figure 3A, 3C, 3D) was markedly decreased in the HCT116-miR-148a cells, but not in the HCT116 + bevacizumab cells (HIF-1α: P = 0.41 and Mcl-1: P = 0.05; Figure 3A, 3C, 3D).
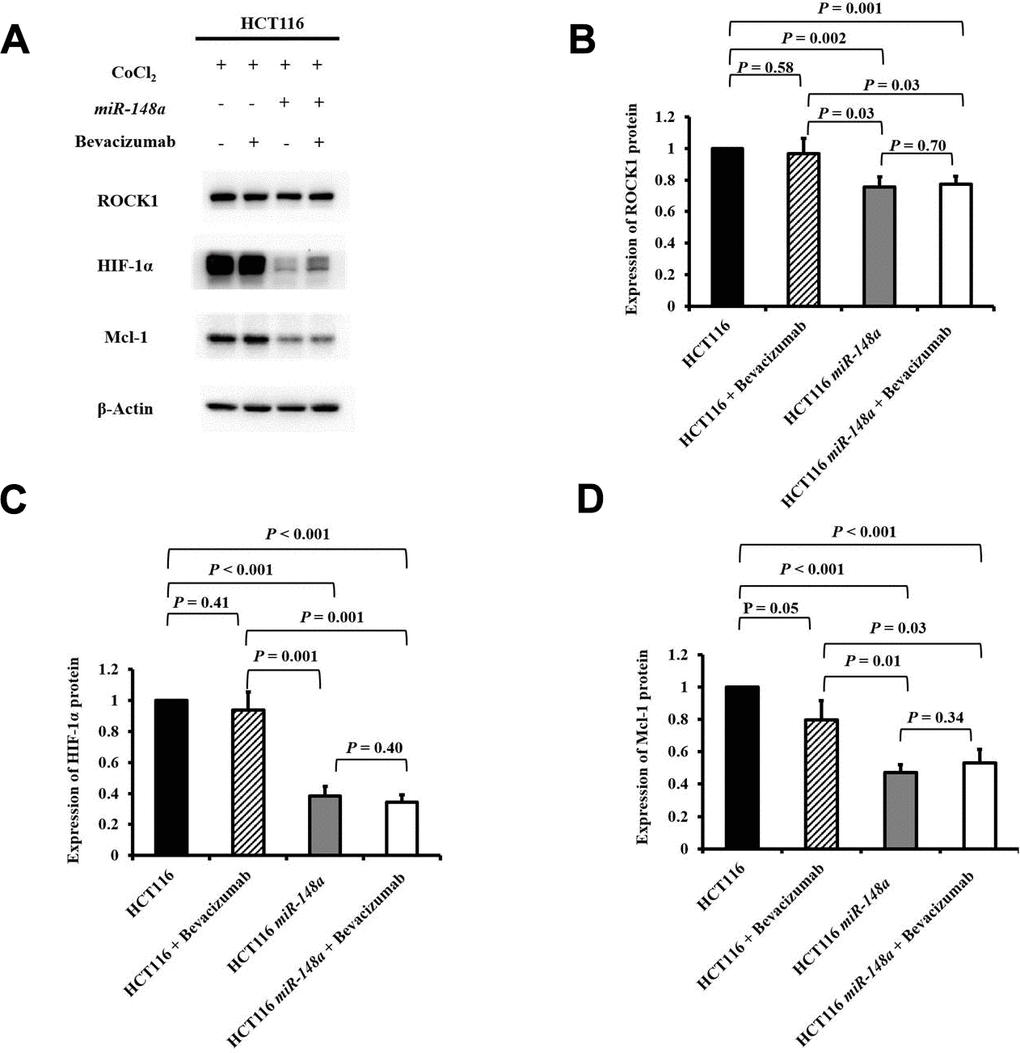
Figure 3. miR-148a inhibited the expression of HIF-1α and Mcl-1 proteins by directly targeting ROCK1 in HCT116 cells under hypoxia. The protein expression levels of ROCK1, HIF-1α, and Mcl-1 were evaluated under a hypoxic condition generated using CoCl2 and in four cell lines (HCT116, HCT116 + bevacizumab, HCT116-miR-148a, and HCT116-miR-148a + bevacizumab). β-Actin served as an internal control. (A) Protein levels of ROCK1, HIF-1α, and Mcl-1; (B) The protein expression level of ROCK1 was significantly decreased in the HCT116-miR-148a cells but not in the HCT116 cells (P = 0.002); (C) The protein expression level of HIF-1α was significantly decreased (P < 0.001); (D) The Mcl-1 protein expression level was markedly decreased (P < 0.001).
miR-148a directly targets Met in the HT29 cell line to curb the expression of HIF-1α and Mcl-1 under hypoxia
To research the effect of miR-148a on the activation of Met, HIF-1α, and Mcl-1 under hypoxia, we further studied the protein expression of Met, HIF-1α, and Mcl-1 in the HT29, HT29 + bevacizumab, HT29-miR-148a, and HT29-miR-148a + bevacizumab cell lines separately. The protein expression of Met was significantly decreased in the HT29-miR-148a cells compared that in with the HT29 cells (Figure 4A, 4B, P < 0.001), but not in the HT29 + bevacizumab (P = 0.28, Figure 4A, 4B). Similarly, the protein expression of HIF-1α and Mcl-1 (all P < 0.001; Figure 4A, 4C, 4D) was significantly decreased in the HT29-miR-148a cells, but not in the HCT29 + bevacizumab cells (HIF-1α: P = 0.09 and Mcl-1: P = 0.05; Figure 4A, 4C, 4D).
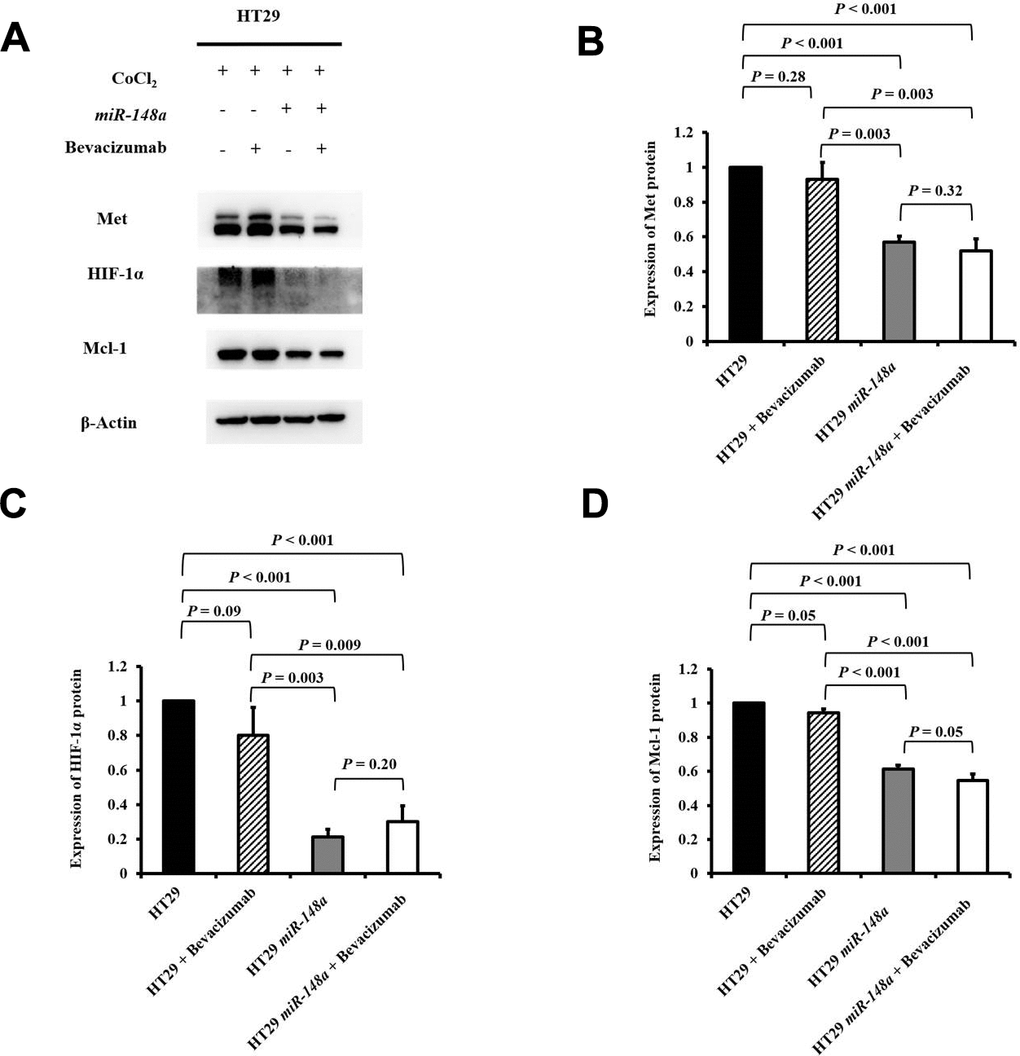
Figure 4. miR-148a inhibited the protein expression of HIF-1α and Mcl-1 by directly targeting Met in the HT29 cell line under hypoxia. We created a hypoxic condition by using CoCl2 and evaluated the expression levels of HIF-1α and Mcl-1 in four cell lines (HT29, HT29 + bevacizumab, HT29-miR-148a, and HT29-miR-148a + bevacizumab). β-Actin served as an internal control. (A) Protein levels of Met, HIF-1α, and Mcl-1; (B) The Met protein expression level was significantly decreased in HT29-miR-148a cells but not in HT29 cells (P < 0.001); (C) The HIF-1α protein expression level was significantly decreased (P < 0.001). (D) The Mcl-1 protein expression level was markedly decreased (P < 0.001).
miR-148a suppressed VEGF secretion and angiogenesis in HCT116 and HT29 colon cancer cells under hypoxia
For the nonhypoxic and hypoxic culture condition, we discovered that miR-148a uncommonly inhibited the expression of HIF-1α and VEGF in HCT116 cells (nonhypoxic: P = 0.028 and 0.0005; hypoxic: P = 0.0007 and 0.02 respectively; Supplementary Figure 2A) and in HT29 cells (nonhypoxic: P = 0.0006 and 0.0014; hypoxic: P = 0.045 and 0.02, respectively; Supplementary Figure 2B). The antiangiogenesis ability of miR-148a was investigated through a tube formation assay in human umbilical vein endothelial cells (HUVECs), and the inhibition of VEGF secretion was assessed through Western blotting. Tube formation was significantly inhibited in the HCT116 cells + bevacizumab group (P = 0.001, Figure 5A, 5B), HT29 cells + bevacizumab group (P < 0.001, Figure 6A, 6B), HCT116-miR-148a cells (P < 0.001, Figure 5A, 5B), and HT29-miR-148a cells (P < 0.001, Figure 6A, 6B). Moreover, miR-148a significantly suppressed VEGF secretion in the HCT116-miR-148a (P = 0.007, Figure 5C, 5D) and HT29-miR-148a (P = 0.004; Figure 6C, 6D) cells, but bevacizumab did not significantly inhibit VEGF secretion in the HCT116 cells + bevacizumab group (P = 0.78, Figure 5C, 5D) or HT29 cells + bevacizumab group (P = 0.69, Figure 6C, 6D).
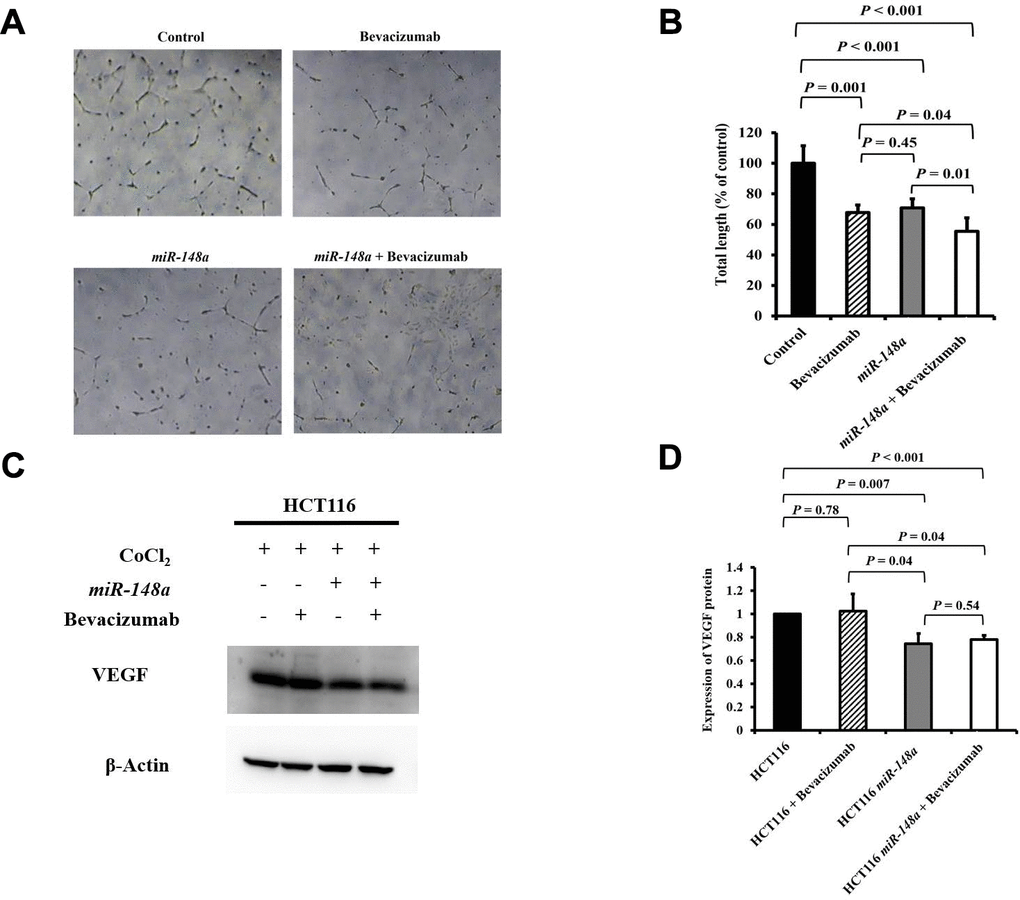
Figure 5. miR-148a suppresses VEGF secretion and the angiogenesis of bevacizumab in HCT116 colon cancer cells under hypoxic conditions. (A) Human umbilical vein endothelial cell tube formation assay was performed in four HCT116 cell lines (HCT116, HCT116 + bevacizumab, HCT116-miR-148a, and HCT116-miR-148a + bevacizumab); (B) Both miR-148a and bevacizumab significantly inhibited human umbilical vein endothelial cell formation (P < 0.001 and P = 0.001; respectively); (C) VEGF expression levels in HCT116, HCT116 + bevacizumab, HCT116-miR-148a, HCT116-miR-148a + bevacizumab cells obtained using Western blotting under hypoxia; (D) miR-148a significantly inhibited VEGF secretion as shown in Western blotting analysis (P = 0.007) but not in HCT116 + bevacizumab cells. β-Actin served as an internal control.
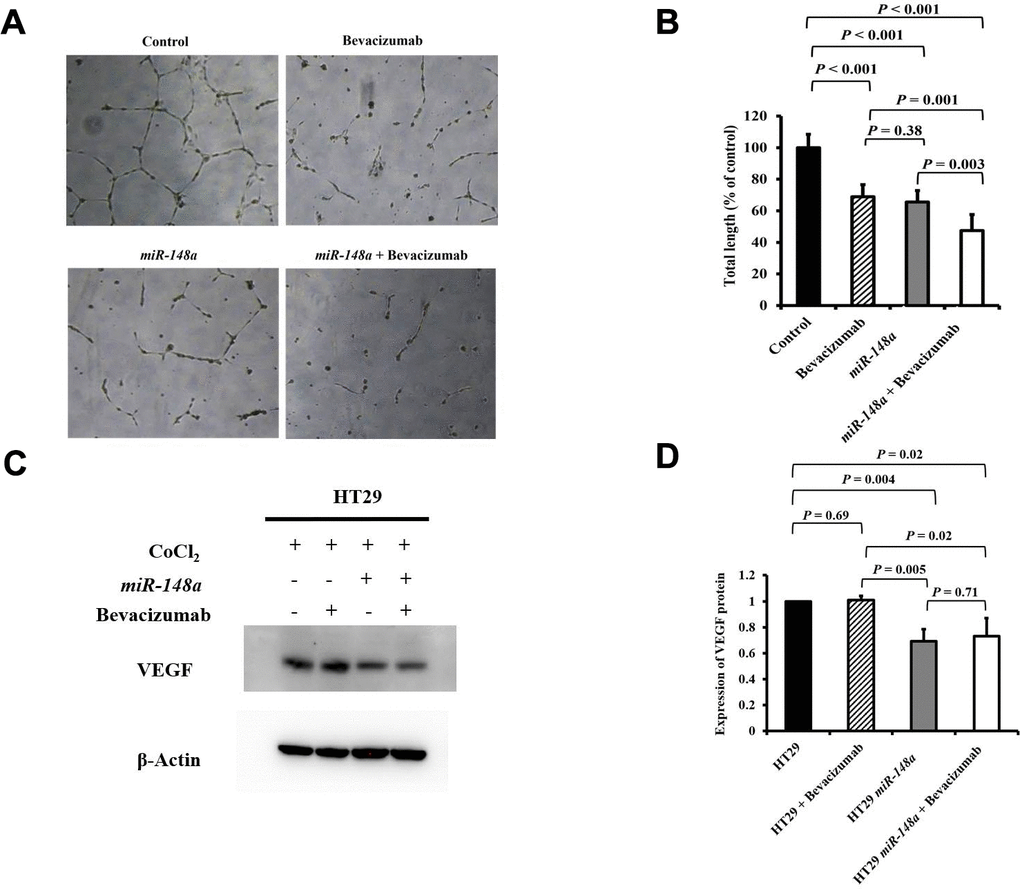
Figure 6. miR-148a suppresses VEGF secretion and the angiogenesis of bevacizumab in HT29 colon cancer cells under hypoxic conditions. (A) Human umbilical vein endothelial cell tube formation assay in four HT29 cell lines (HT29, HT29 + bevacizumab, HT29-miR-148a, and HT29-miR-148a + bevacizumab); (B) Both miR-148a and bevacizumab significantly inhibited human umbilical vein endothelial cell formation (all P < 0.001); (C) VEGF expression levels in HT29, HT29 + bevacizumab, HT29-miR-148a, and HT29-miR-148a + bevacizumab as obtained using Western blotting under hypoxia; (D) miR-148a significantly inhibited VEGF secretion as shown in the Western blotting analysis (P = 0.004) but not in HT29 + bevacizumab cells. β-Actin served as an internal control.
miR-148a overexpression inhibited the viability of HCT116 and HT29 cells
miR-148a remarkably inhibited cell viability regardless of the concentrations of bevacizumab at 24, 48, and 72 h (all P < 0.001; Figure 7). This result indicated the miR-148a alone could directly suppress the proliferation of colon cancer cells.
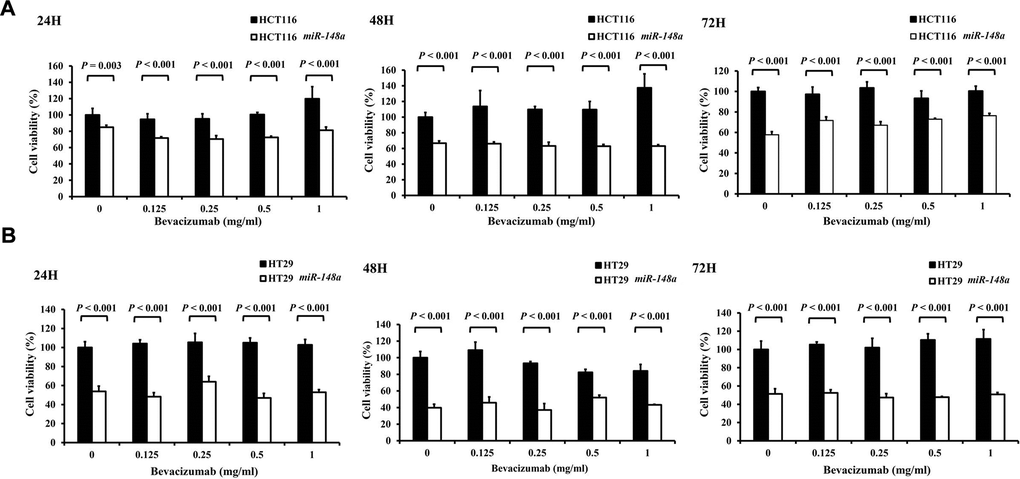
Figure 7. The anti-cell viability effect of miR-148a on HCT116 and HT-29 colon cancer cells. Four cell lines (HCT116, HCT116-miR148a, HT29, and HT29-miR-148a) were treated with different concentrations of bevacizumab for 24, 48, and 72 h. (A) HCT116 and HCT116-miR148a cells; (B) HT29 and HT29-miR-148a cells. miR-148a significantly inhibited cell viability regardless of the concentration of bevacizumab at 24, 48, and 72 h (all P < 0.001).
Synergistic antitumorigenic effect of miR-148a and bevacizumab on nude mice
One week after implantation, mice were assigned to two groups, and saline (Left; Figure 8A) or bevacizumab (Right; Figure 8A) was injected at the tumor site to evaluate the synergistic antitumorigenic effect. Mice that received bevacizumab had significantly smaller cancer lumps (P = 0.005; Figure 8B, 8C) and lower tumor weight (P = 0.02; Figure 8D) than did those that received saline. Furthermore, the coadministration of miR-148a and bevacizumab exerted significant synergistic effects on the reduction of tumor volume (P = 0.007, Figure 8C) and tumor weight (P = 0.004, Figure 8D). The results suggest that similar to the effect of bevacizumab, miR-148a overexpression leads to a prominent reduction in tumor cell proliferation in animal models.
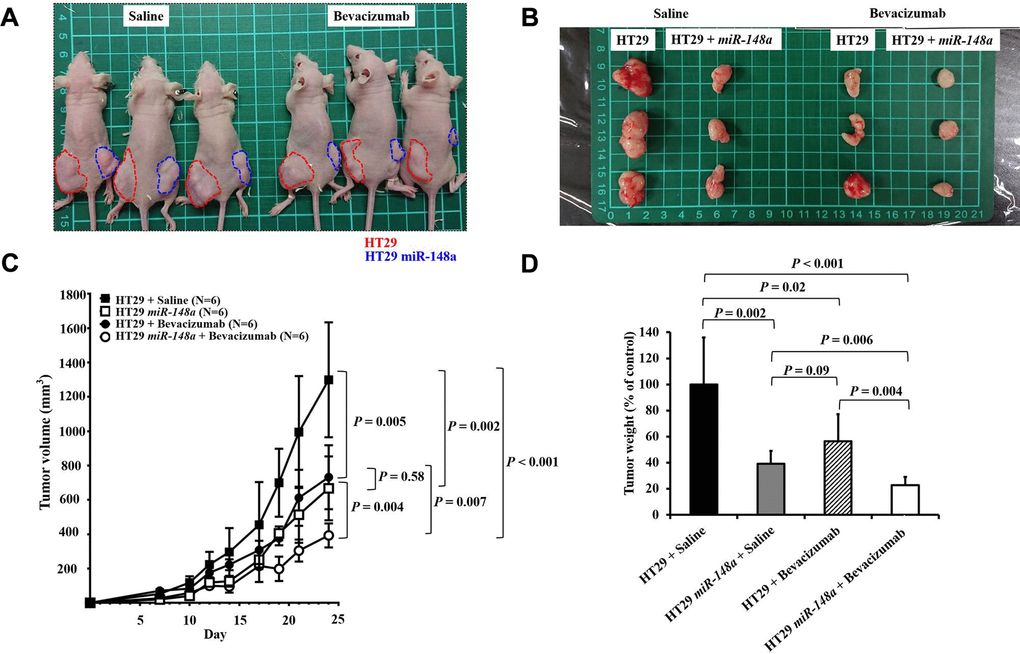
Figure 8. Synergistic anti-tumorigenic effect in animal study. To validate the role of miR-148a in tumorigenesis and evaluate the effect of miR-148a overexpression on tumor growth in vivo, miR-148a overexpression and NC clones with scrambled pCDH-NC were injected subcutaneously in 8-week-old nude mice to allow tumor growth (red circle: NC; blue circle: miR-148a overexpression). (A) One week after implantation, the mice were assigned to two groups and saline (Left) or bevacizumab (Right) was injected at the tumor site to evaluate the synergistic anti-tumorigenic effect. (B) Mice that received bevacizumab and miR-148a overexpression had significantly smaller cancer lumps than those that received saline and NC. (C) After the tumor-bearing mice were sacrificed at 3 weeks after tumor cell seeding, tumor burdens were analyzed. Mice that received bevacizumab had significantly smaller cancer lumps than those that received saline (P = 0.005) but larger lumps than those that received bevacizumab + miR-148a overexpression (P = 0.007). (D) Mice that received bevacizumab had significantly lower tumor weight than those that received saline (P = 0.02) but higher tumor weight than those that received bevacizumab + miR-148a overexpression (P = 0.004).
Association between serum expression of miR-148a and therapeutic response in mCRC patients
To evaluate the role of miR-148a expression in mCRC patients treated with bevacizumab, we collected the serum samples of 24 mCRC patients before they received bevacizumab plus FOLFIRI as the first-line treatment. Among these patients, 14 showed a partial response (PR), and 10 had progressive disease (PD) (Figure 9A). The patients who exhibited PR had significantly higher serum miR-148a expression than those who had PD (P = 0.026, Figure 9B). This result indicated that mCRC patients who exhibited miR-148a overexpression receiving bevacizumab plus FOLFIRI had a more favorable treatment response than those with lower miR-148a expression.
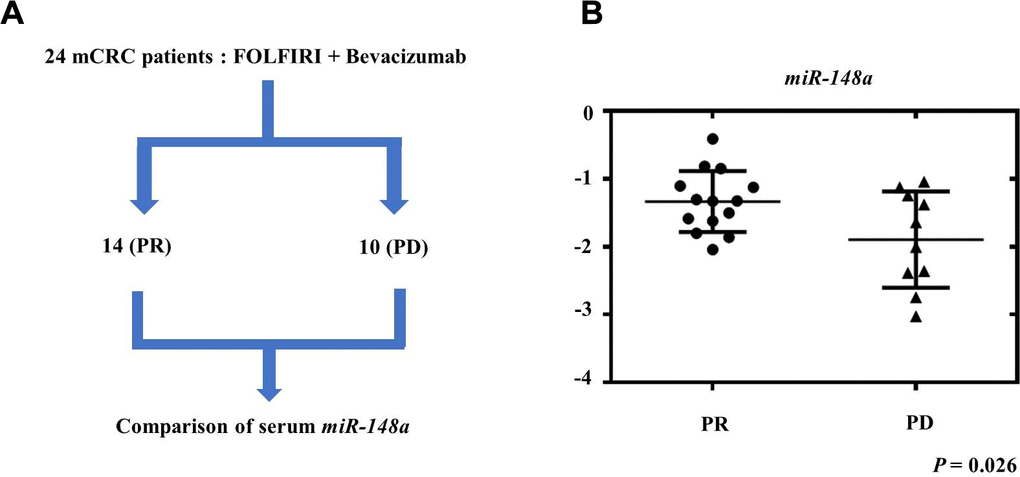
Figure 9. Relationship between therapeutic response and serum miR-148a expression in mCRC patients. We collected the serum samples of 24 mCRC patients before they received treatment with bevacizumab plus FOLFIRI as first-line regimen. (A) After treatment, 14 patients showed partial response (PR) and 10 patients had progressive disease (PD). (B) miR-148a expression was significantly higher in the serum samples of the 14 PR patients than in those of the 10 PD patients (P = 0.026).
Discussion
One of the main findings of the current study is that miR-148a could decrease angiogenesis in and increase the apoptosis of colon cancer cells via direct downregulation of ROCK1 and c-Met and their relevant pathways. Even under hypoxic conditions, miR-148a efficiently inhibited the expression of HIF-1α, VEGF, and Mcl-1. We re-verified that miR-148a can inhibit angiogenesis in and decrease the viability of CRC cells whether it is in vitro or in vivo conditions. Moreover, a notable synergistic effect of miR-148a and bevacizumab on antitumorigenesis was observed in the animal model.
ROCK1 is an essential effector kinase of Rho GTPases and plays a vital role in regulating tumor invasion and metastasis [25, 26]. In 2011, Zheng et al. demonstrated that miR-148a suppressed tumor cell invasion and metastasis by downregulating ROCK1 in gastric cancer cells [29]. c-Met, a tyrosine receptor, plays a key role in the EMT and metastasis in hepatocellular carcinoma [27, 28]. In 2014, Zhang et al. reported that miR-148a may inhibit Met/Snail signaling and may negatively regulate the EMT and metastasis of hepatoma cells [17]. We confirmed that miR-148a directly targeted the 3′-UTR of ROCK1 and c-Met in colon cancer cell lines in the current study.
Angiogenesis is a complicated process in which the formation of new blood vessels are from an endothelial precursor in malignancies [6]. This process is tightly regulated and mediated by a group of ligands [30, 31]. Both HIF-1α and VEGF play critical roles in angiogenesis and tumor progression [32, 33]. The process of programmed cell death and a homeostatic mechanism that helps maintain cell populations during normal cell development and aging is called apoptosis. It maintains balance between the generation of new cells and the loss of cells in normal tissues. Impaired apoptosis is an important step in tumorigenesis. Apoptosis has two underlying signaling pathways: extrinsic and intrinsic [34, 35]. miR-148a deactivates the intrinsic mitochondrial pathway via Bcl-2 inhibition and tumor apoptosis induction in CRC [8, 16]. Mcl-1, a prosurvival protein of the Bcl-2 family, exhibits anti-apoptotic ability. It is the main regulator of cell death [36] and targets the phosphorylation of the protein kinase Raf-1 to inactivate cell death signaling pathways [37, 38].
Many studies have revealed the role of miR-148a in multiple malignancies, including in inhibiting the growth of pancreatic and prostate cancer cells [39, 40] and suppressing angiogenesis in breast cancer [41]. We previously demonstrated that miR-148a suppressed VEGF through the downregulation of the pERK/HIF-1α/VEGF pathway, thereby inhibiting angiogenesis [7]. The overexpression of miR-148a assisted apoptosis and curbed proliferation by directly targeting c-Met in vitro and improved the tumor response to irradiation in vivo [19]. Nersisyan et al. reported that the ITGA5 gene and PRNP gene were upregulated in Caco-2 cells exposed to chemical hypoxia. The Cancer Genome Atlas Colon Adenocarcinoma cohort reported that elevated expression levels of both these genes were associated with poor prognosis in patients with CRC. Further, miR-148a may directly interact with both genes to influence tumor progression and metastasis [42]. Thus, miR-148a functions as a potential tumor suppressor through diverse mechanisms. In this study, we affirmed that miR-148a indirectly inhibited the expression of HIF-1α and Mcl-1 by directly binding to ROCK1 and c-Met, thereby enhancing apoptosis of colon cancer cells and decreasing angiogenesis in tissues containing such cells. Notably, we also found that bevacizumab acts as an angiogenesis inhibitor by inhibiting VEGF-A and arresting tumor growth. However, it could not inhibit VEGF secretion. Nevertheless, whether bevacizumab leads to tumor cell apoptosis through the downregulation of Mcl-1 secretion remains uncertain. The overexpression of miR-148a synergistically enhanced the antitumorigenic and apoptotic effects of bevacizumab, thereby yielding better therapeutic outcomes than bevacizumab-only treatment in vitro and in vivo.
The study has some limitations. First, the sample size was relatively small. Future studies should enroll a larger cohort to confirm the predictive value of miR-148a in patients with mCRC. Second, the synergistic effects of miR-148a and bevacizumab on tumor volume and weight reduction could be demonstrated in the animal model, but not in colon cancer cell lines. Third, the evidence regarding ROCK1/c-Met, HIF-1α/VEGF, and Mcl-1 in this study is based on the results of other studies. This finding should be validated in future investigations.
Finally, our research demonstrated that miR-148a downregulated HIF-1α/VEGF and Mcl-1 by directly targeting ROCK1/c-Met to decrease angiogenesis and increase the apoptosis of colon cancer cells. Clinically, we demonstrated that patients with mCRC with serum miR-148a overexpression have more a favorable therapeutic response than those undergoing the combination therapy of chemotherapy and bevacizumab (standard dose; 5 mg/kg). Therefore, the miR-148a status possesses prognostic/predictive value in patients with advanced CRC treated with anti-VEGF biological agents and has clinical implications in improving therapeutic strategies and designing personalized treatment for this malignancy.
Materials and Methods
Study design
We demonstrated the relationship between miR-148a and VEGF indirect downregulation by targeting HIF-1α under nonhypoxic and hypoxic conditions in our previous study [7]. Herein, the TargetScan program (https://targetscan.org) and the isomiRTar portal (https://isomirtar.hse.ru/) were made use of identifying potential genes which miR-148a directly targets. We only considered the conserved genes which carried conserved sequences. We used Gene Ontology (http://geneontology.org) software to discover the function of miR-148a target genes. A previous bioinformatics analysis of pathways revealed that miR-148a directly targets ROCK1 and c-Met, thereby affecting the function of Mcl-1 [43]. Accordingly, we hypothesized that miR-148a inhibits the secretion of VEGF and Mcl-1 proteins by directly targeting ROCK1 and c-Met and can therefore enhance the apoptosis of and downregulate angiogenesis in cancer cells (Figure 10A). The design of the cell-line study is presented in Figure 10B, and the process of the animal study is detailed in Supplementary Figure 3.
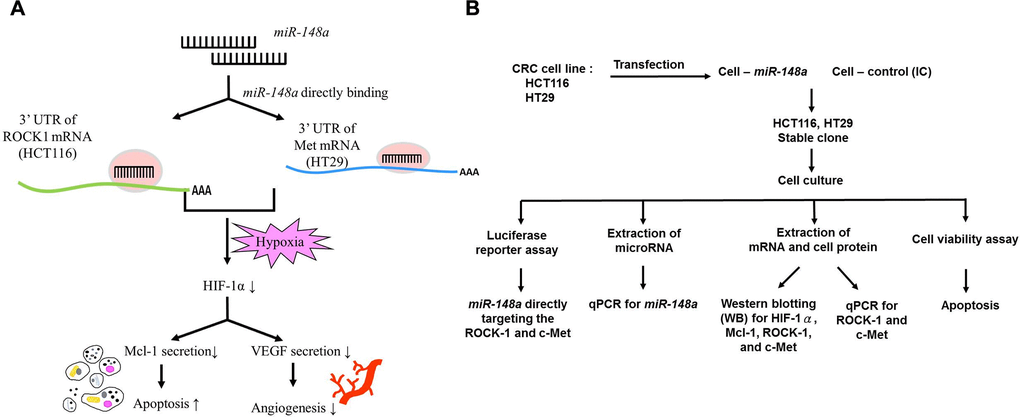
Figure 10. Study hypothesis and design. (A) We hypothesized that miR-148a inhibits the secretion of VEGF and Mcl-1 through the inactivation of HIF-1α by directly targeting ROCK1 and c-Met. This induced the apoptosis of and reduced angiogenesis in cancer cells. (B) In vitro, we transfected miR-148a into HCT116 and HT29 cells and established stable CRC clones. The luciferase reporter assay was performed to prove the direct targeting of ROCK1 and c-Met by miR-148a. The protein levels of HIF-1α, Mcl-1, ROCK1, and Met were examined through Western blotting, and the mRNA levels of ROCK1 and c-Met were tested through RT-PCR. The cell viability assay was used to examine the apoptosis.
Cell lines and cell line authentication
Five colon cancer cell lines—HCT116, HT29, SW480, SW620, and Caco-2—were used for the transfection and proliferation of miR-148a; the HCT116 and HT29 cells exhibited higher fold changes and relative proliferation after transfection than the other cell lines (Supplementary Figure 4A, 4B). In addition, the activation of the RAS-RAF pathway was related to increase VEGF-induced angiogenesis. We also evaluated the role of miR-148a in and the effects of BRAF mutations (HT29 cells) and KRAS mutations (HCT116 cells) on angiogenesis. We purchased the HCT116 and HT29 cells from the Bioresource Collection and Research Center (Hsinchu, Taiwan) and American Type Culture Collection (Manassas, VA, USA), respectively. We cultured the all of cell lines in Dulbecco's modified Eagle’s medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco), 100 IU/mL penicillin (Gibco), and 100 μg/mL streptomycin (Gibco) in a humid atmosphere containing 5% CO2 at 37° C. HUVECs, a part of the Angiogenesis Starter Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA), was also assayed.
We resuscitated the HCT116 and HT29 cell lines and cultured them for 2 weeks after delivery from the provider and were then transfected with the pCDH vector (System Biosciences, Palo Alto, CA, USA) expressing miR-148a. We used the transfected cell lines in subsequent experiments. Through transduction in the target cells, the pCDH expression vector could integrate into genomic DNA, thus providing stable, long-term expression of the target gene. The genome of the transfected cells was different from that of the wild-type cells because of the genomic integration of the expression vector. Therefore, the pCDH-transfected cells were no longer identified by their original genomic status compared with the wild-type cells in the cell line authentication test. Because the genomic changes were expected, the cell lines used in this study were not subjected to an authentication test.
Patient tissue samples
To assess the correlation between the effect of bevacizumab (Avastin, Roche, Basel, Switzerland) treatment and miR-148a expression levels, patients with mCRC who received FOLFIRI plus bevacizumab (Avastin) were classified into two groups according to their treatment response: The PR cohort, which comprised 14 patients, and the PD cohort, which comprised 10 patients. The serum samples of the patients in the two cohorts were collected for RNA extraction. All clinical samples were collected after written informed consent was obtained from each participant. The study protocol was approved by the Institutional Review Board of Kaohsiung Medical University Hospital (KMUHIRB-G(II)20190039).
Establishment of vectors with overexpressed miR-148a
To evaluate the functional effects of overexpressed miR-148a, the pCDH vector (System Bioscience, Mountain View, CA, USA) was used as the overexpressed miR-148a system. We constructed the pCDH-miR-148a plasmid by intercalating the miR-148a PCR product into multiple cloning sites of the pCDH vector. The following PCR primer sequences were used for miR-148a cloning: GCCTGAATTCATGCTTTTAACGAGTTATTCTTC and CTAGGCGGCCGCGCCTTGCCCCTCCCCCAAGGA. The forward and reverse primers were extended by inclusion of GAATTC and GCGGCCGC sequences, respectively, creating EcoR1 and Not1 restriction sites at their 5′ end, respectively. The miR-148a overexpression vectors were confirmed through direct DNA sequencing.
Establishing stable clones
The HCT116 and HT29 cells (5 × 105) were seeded and transfected them with the negative-scrambled pCDH vector or the pCDH–miR-148a plasmid (400 ng) by using Lipofectamine 3000 (Thermo Fisher Scientific). The transfected cells were cultured in standard culture media supplemented with 12 μg/mL puromycin (Thermo Fisher Scientific) over 4 weeks to achieve pCDH-negative or pCDH–miR-148a stable clone selection. The TaqMan miRNA quantitative PCR (RT-qPCR) assay (Applied Biosystems, Waltham, MA, USA) was conducted to confirm the stable expression of the transfected plasmid.
Luciferase reporter assay
To verify miR-148a-directed binding on targeted mRNA, two 3′-UTR segments including one wild-type and the other mutant type were cloned and integrated into the pMirTarget Vector (OriGene Technologies, Inc., Rockville, MD, USA), respectively. Both the wild-type and mutant 3′-UTR segments of c-Met or ROCK1 were constructed, because c-Met and ROCK1 were predicted to be the potential target genes of miR-148a. The cells (1 × 104) were seeded in a 96-well plate for 24 h and co-transfected with two plasmids—wild-type or mutant 3′-UTR construct and pTK-Green Renilla Luc Vector (Thermo Fisher Scientific)—by using Lipofectamine 3000 (Thermo Fisher Scientific). After transfection for 48 h, a luciferase reporter assay was performed using the Pierce™ Renilla-Firefly Luciferase Dual Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. For the normalization of firefly activity, the Renilla luciferase activity was used as the internal control.
RNA extraction and cDNA preparation
Approximately 107 cells were prepared for the extraction of RNA including mRNA and miR. Total RNA purification was performed using Qiagen RNAeasy Columns (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions. To synthesize the cDNA of miR-148a or U6 for the detection of miR-148a expression levels, 100 ng of total RNA with the unique primer (Applied Biosystems) was used, and to synthesize the cDNA of mRNA for studying the mRNA expression level of miR-148a-mediated genes, 2 μg of total RNA with random hexamer primers (Applied Biosystems) was applied.
miR-148a expression levels in patient samples and CRC cell lines
The TaqMan miR RT-qPCR assay (Applied Biosystems) was applied to quantify the miR-148a expression level. RT-qPCR was performed using the Applied Biosystems 7900HT Real-Time PCR System. The relative miR-148a expression level in each serum sample was normalized to the internal control of U6 snRNA in accordance with the following equation: log10(2−ΔCt), where ΔCt = (CtmiR-148a − CtU6).
mRNA expression level
The mRNA expression levels of miR-148a-mediated genes were quantified with SYBR Green (Applied Biosystems) by using the Applied Biosystems 7900HT Real-Time PCR System. The relative expression level in each sample was normalized to the internal control of glyceraldehyde 3-phosphate dehydrogenase and was evaluated using the following formula: (2−ΔΔCt).
HUVEC tube formation assay
Angiogenesis was induced in vitro by using the Angiogenesis Starter Kit (Gibco, Grand Island, NY, USA) according to the manufacturer’s manual. In brief, a day before the tube formation assay was performed, the Geltrex Matrix was thawed at 4° C and then bottom coated in a 24-well plate at 37° C for 30 min. Subsequently, the HUVECs were seeded on the Geltrex Matrix–coated plate and incubated for 24 h, after which the conditional medium collected from transfected cells was added to each well. Following 24 h of incubation, cell images were taken using the Edipse Ti-U inverted microscope system (Nikon, Inc., Melville, NY, USA), and the total length of the tube formed was measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Cell viability assay
To study the anticancer effects of miR-148a, we first treated the HCT116, HCT116-miR-148a, HT29, and HT29-miR-148a cells with various concentrations (0 mg/ml, 0.125 mg/ml, 0.25 mg/ml, 0.5 mg/ml, and 1 mg/ml) of bevacizumab for 24, 48, and 72 h, respectively, and evaluated cell viability by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay kit (Sigma-Aldrich, St. Louis, MO, USA). We seeded the cells (5 × 103 cells/well) into a 96-well culture plate and incubated the plate overnight at 37° C before treatment with the appropriate dose of bevacizumab (Avastin) (0, 0.125, 0.25, 0.5, or 1 mg/mL) (Roche). After 24-, 48-, and 72-h incubation, 20 μL of 5 mg/mL MTT was added to each well and incubated at 37° C for 2 h. The medium was replaced with 100 μL of dimethyl sulfoxide to dissolve the precipitate. Thereafter, absorbance was measured at 570 nm on a 96-well microplate reader (BioTeK Instruments, Winooski, VT, USA).
Western blotting and antibodies
All the cells were harvested and lysed with ice-cold RIPA buffer (Merck Millipore, Burlington, MA, USA), protease inhibitor cocktail (Sigma-Aldrich), and phosphatase inhibitor cocktail (Sigma-Aldrich). Equal amounts (30 μg) of protein were resolved through sodium dodecyl sulfate-polyacrylamide gel electrophoresis and were transferred onto polyvinylidene difluoride (PVDF) membranes (Merck Millipore). After blocking with 5% skim milk for 1 h, the PVDF membranes were incubated with primary antibodies, such as anti-VEGF 165A (Abcam PLC, Cambridge, England, UK), anti-ROCK1 (Abcam PLC, Cambridge, England, UK), anti-Met (Cell Signaling Technology, Danvers, MA, USA), anti-HIF-1α (Cell Signaling Technology), anti-Mcl-1 (Cell Signaling Technology), and anti-β-Actin (Sigma-Aldrich), overnight at 4° C. Following washing with tris-buffered saline containing Tween 20 (TBS-Tween 20), the PVDF membranes were incubated with the secondary antibody at room temperature for 1 h. After the membranes were washed, immunoreactive proteins were detected using a SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Inhibition of HIF-1α and VEGF and Mcl-1 expression by miR-148a under hypoxia
Seo et al. [44] demonstrated CoCl2 to be a hypoxia mimetic agent. Therefore, we used CoCl2 to create a hypoxic culture condition and demonstrated the ability of miR-148a to inhibit HIF-1α, VEGF, and Mcl-1 expression under such a condition.
Xenograft study
We further validated the role of miR-148a in anti-tumorigenesis and evaluated the effect of miR-148a overexpression on tumor growth in vivo. Six-week-old male Balb/c nude mice were purchased from BioLASCO Taiwan, Co., Ltd. (Taipei, Taiwan) and were maintained in the Center for Laboratory Animals of Kaohsiung Medical University. At 8 weeks of age, the mice were subcutaneously injected with miR-148a overexpression and NC clones with scrambled pCDH-NC for tumor growth (red circle: NC; blue circle: miR-148a overexpression). Each mouse received two subcutaneous injections of 1 × 107 HT29 cells (either pCDH-negative or pCDH–miR-148a) into the bilateral flank for the implantation of two tumors. One week after implantation, the mice were assigned into two groups—saline only or bevacizumab. The mice received an intraperitoneal injection of bevacizumab (2.5 mg/kg) (Roche) or an equal volume of saline twice per week. The tumor diameter was measured, and tumor volume was calculated using the following formula: V = (length × width2)/2. After the tumor-bearing mice were sacrificed at 3 weeks after tumor cell seeding, tumor burdens were analyzed. All animal experiments were performed in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited facility. The animal handling procedures were in accordance with the protocols approved by the Institutional Animal Care and Use Committee of Kaohsiung Medical University (IACUC Approval No: 108165).
Statistical analysis
All statistical analyses were performed using SPSS V21.0 (IBM SPSS, Chicago, IL, USA). Data are expressed as the mean ± standard deviation values of at least three independent experiments. Significant differences between two groups were determined using Student’s t-test analyses. A P value of <0.05 was considered statistically significant.
Supplementary Materials
Author Contributions
Tsai HL, Tsai YC, and Wang JY participated in the study design, molecular genetic studies, and carried out the sequence alignment. Tsai HL drafted and wrote the manuscript. Tsai YC was responsible for molecular genetic studies. Wang JY conceived the study and participated in its design and coordination. Chen YC, Huang CW, Chen PJ, Li CC, Su WC, Chang TK, Yeh YS, and Yin TC were responsible for data collection and analysis. All authors have read and approved the final manuscript.
Acknowledgments
This work was supported by grants through funding from the Ministry of Science and Technology (MOST 109-2314-B-037-046-MY3, MOST110-2314-B-037-097, MOST 111-2314-B-037-070-MY3, MOST 111-2314-B-037-049) and the Ministry of Health and Welfare (MOHW111-TDU-B-221-114014) and funded by the health and welfare surcharge of on tobacco products, and the Kaohsiung Medical University Hospital (KMUH110-0R37, KMUH110-0R38, KMUH110-0M34, KMUH110-0M35, KMUH110-0M36, KMUH-DK(B)110004-3) and KMU Center for Cancer Research (KMU-TC111A04-1) and KMU Office for Industry-Academic Collaboration (S109036), Kaohsiung Medical University. In addition, this study was supported by the Grant of Taiwan Precision Medicine Initiative and Taiwan Biobank, Academia Sinica, Taiwan, R.O.C.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Ethical Statement and Consent
All animal experiments were performed in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited facility. The animal handling procedures were in accordance with the protocols approved by the Institutional Animal Care and Use Committee of Kaohsiung Medical University (IACUC Approval No: 108165).
All human clinical samples were collected after written informed consent was obtained from each participant. The study protocol was approved by the Institutional Review Board of Kaohsiung Medical University Hospital (KMUHIRB-G(II)20190039).
Funding
This study was funded in part by the Ministry of Science and Technology, the Ministry of Health and Welfare, Taiwan Precision Medicine Initiative, Academia Sinica, Taiwan, and the Kaohsiung Medical University Hospital, Kaohsiung Medical University.
References
- 1. Peng YC, Lin CL, Hsu WY, Chang CS, Yeh HZ, Liao SC, Kao CH. The risk of colorectal cancer is related to frequent hospitalization of IBD in an Asian population: results from a nationwide study. QJM. 2015; 108:457–63. https://doi.org/10.1093/qjmed/hcu225 [PubMed]
- 2. Hibino Y, Sakamoto N, Naito Y, Goto K, Oo HZ, Sentani K, Hinoi T, Ohdan H, Oue N, Yasui W. Significance of miR-148a in Colorectal Neoplasia: Downregulation of miR-148a Contributes to the Carcinogenesis and Cell Invasion of Colorectal Cancer. Pathobiology. 2015; 82:233–41. https://doi.org/10.1159/000438826 [PubMed]
- 3. Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019; 14:89–103. https://doi.org/10.5114/pg.2018.81072 [PubMed]
- 4. Tsai HL, Chen YC, Yin TC, Su WC, Chen PJ, Chang TK, Li CC, Huang CW, Wang JY. Comparison of UGT1A1 Polymorphism as Guidance of Irinotecan Dose Escalation in RAS Wild-Type Metastatic Colorectal Cancer Patients Treated With Cetuximab or Bevacizumab Plus FOLFIRI as the First-Line Therapy. Oncol Res. 2022; 29:47–61. https://doi.org/10.3727/096504022X16451187313084 [PubMed]
- 5. Takahashi M, Cuatrecasas M, Balaguer F, Hur K, Toiyama Y, Castells A, Boland CR, Goel A. The clinical significance of MiR-148a as a predictive biomarker in patients with advanced colorectal cancer. PLoS One. 2012; 7:e46684. https://doi.org/10.1371/journal.pone.0046684 [PubMed]
- 6. Mousa L, Salem ME, Mikhail S. Biomarkers of Angiogenesis in Colorectal Cancer. Biomark Cancer. 2015 (Suppl 1); 7:13–9. https://doi.org/10.4137/BIC.S25250 [PubMed]
- 7. Tsai HL, Miao ZF, Chen YT, Huang CW, Yeh YS, Yang IP, Wang JY. miR-148a inhibits early relapsed colorectal cancers and the secretion of VEGF by indirectly targeting HIF-1α under non-hypoxia/hypoxia conditions. J Cell Mol Med. 2019; 23:3572–82. https://doi.org/10.1111/jcmm.14257 [PubMed]
- 8. Wang H. MicroRNAs and Apoptosis in Colorectal Cancer. Int J Mol Sci. 2020; 21:5353. https://doi.org/10.3390/ijms21155353 [PubMed]
- 9. Doleshal M, Magotra AA, Choudhury B, Cannon BD, Labourier E, Szafranska AE. Evaluation and validation of total RNA extraction methods for microRNA expression analyses in formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2008; 10:203–11. https://doi.org/10.2353/jmoldx.2008.070153 [PubMed]
- 10. Di Leva G, Croce CM. Roles of small RNAs in tumor formation. Trends Mol Med. 2010; 16:257–67. https://doi.org/10.1016/j.molmed.2010.04.001 [PubMed]
- 11. Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007; 449:682–8. https://doi.org/10.1038/nature06174 [PubMed]
- 12. Yang IP, Tsai HL, Hou MF, Chen KC, Tsai PC, Huang SW, Chou WW, Wang JY, Juo SH. MicroRNA-93 inhibits tumor growth and early relapse of human colorectal cancer by affecting genes involved in the cell cycle. Carcinogenesis. 2012; 33:1522–30. https://doi.org/10.1093/carcin/bgs166 [PubMed]
- 13. Farazi TA, Hoell JI, Morozov P, Tuschl T. MicroRNAs in human cancer. Adv Exp Med Biol. 2013; 774:1–20. https://doi.org/10.1007/978-94-007-5590-1_1 [PubMed]
- 14. Nagaraju GP, Bramhachari PV, Raghu G, El-Rayes BF. Hypoxia inducible factor-1α: Its role in colorectal carcinogenesis and metastasis. Cancer Lett. 2015; 366:11–8. https://doi.org/10.1016/j.canlet.2015.06.005 [PubMed]
- 15. Li Y, Deng X, Zeng X, Peng X. The Role of Mir-148a in Cancer. J Cancer. 2016; 7:1233–41. https://doi.org/10.7150/jca.14616 [PubMed]
- 16. Zhang H, Li Y, Huang Q, Ren X, Hu H, Sheng H, Lai M. MiR-148a promotes apoptosis by targeting Bcl-2 in colorectal cancer. Cell Death Differ. 2011; 18:1702–10. https://doi.org/10.1038/cdd.2011.28 [PubMed]
- 17. Zhang JP, Zeng C, Xu L, Gong J, Fang JH, Zhuang SM. MicroRNA-148a suppresses the epithelial-mesenchymal transition and metastasis of hepatoma cells by targeting Met/Snail signaling. Oncogene. 2014; 33:4069–76. https://doi.org/10.1038/onc.2013.369 [PubMed]
- 18. Tsai HL, Yang IP, Huang CW, Ma CJ, Kuo CH, Lu CY, Juo SH, Wang JY. Clinical significance of microRNA-148a in patients with early relapse of stage II stage and III colorectal cancer after curative resection. Transl Res. 2013; 162:258–68. https://doi.org/10.1016/j.trsl.2013.07.009 [PubMed]
- 19. Huang CM, Huang MY, Chen YC, Chen PJ, Su WC, Chang TK, Li CC, Huang CW, Tsai HL, Wang JY. miRNA-148a Enhances the Treatment Response of Patients with Rectal Cancer to Chemoradiation and Promotes Apoptosis by Directly Targeting c-Met. Biomedicines. 2021; 9:1371. https://doi.org/10.3390/biomedicines9101371 [PubMed]
- 20. Tsai HL, Pang SY, Wang HC, Luo CW, Li QL, Chen TY, Fang SY, Wang JY, Pan MR. Impact of BMI1 expression on the apoptotic effect of paclitaxel in colorectal cancer. Am J Cancer Res. 2019; 9:2544–53. [PubMed]
- 21. Xu Z, Zhu C, Chen C, Zong Y, Feng H, Liu D, Feng W, Zhao J, Lu A. CCL19 suppresses angiogenesis through promoting miR-206 and inhibiting Met/ERK/Elk-1/HIF-1α/VEGF-A pathway in colorectal cancer. Cell Death Dis. 2018; 9:974. https://doi.org/10.1038/s41419-018-1010-2 [PubMed]
- 22. Glück AA, Orlando E, Leiser D, Poliaková M, Nisa L, Quintin A, Gavini J, Stroka DM, Berezowska S, Bubendorf L, Blaukat A, Aebersold DM, Medová M, Zimmer Y. Identification of a MET-eIF4G1 translational regulation axis that controls HIF-1α levels under hypoxia. Oncogene. 2018; 37:4181–96. https://doi.org/10.1038/s41388-018-0256-6 [PubMed]
- 23. Ohta T, Takahashi T, Shibuya T, Amita M, Henmi N, Takahashi K, Kurachi H. Inhibition of the Rho/ROCK pathway enhances the efficacy of cisplatin through the blockage of hypoxia-inducible factor-1α in human ovarian cancer cells. Cancer Biol Ther. 2012; 13:25–33. https://doi.org/10.4161/cbt.13.1.18440 [PubMed]
- 24. Wu F, Tong DD, Ni L, Wang LM, Wang MC. HIF-1α suppresses myeloma progression by targeting Mcl-1. Int J Clin Exp Pathol. 2020; 13:1483–91. [PubMed]
- 25. Pinner S, Sahai E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol. 2008; 10:127–37. https://doi.org/10.1038/ncb1675 [PubMed]
- 26. Narumiya S, Tanji M, Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009; 28:65–76. https://doi.org/10.1007/s10555-008-9170-7 [PubMed]
- 27. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012; 12:89–103. https://doi.org/10.1038/nrc3205 [PubMed]
- 28. Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000; 2:76–83. https://doi.org/10.1038/35000025 [PubMed]
- 29. Zheng B, Liang L, Wang C, Huang S, Cao X, Zha R, Liu L, Jia D, Tian Q, Wu J, Ye Y, Wang Q, Long Z, et al. MicroRNA-148a suppresses tumor cell invasion and metastasis by downregulating ROCK1 in gastric cancer. Clin Cancer Res. 2011; 17:7574–83. https://doi.org/10.1158/1078-0432.CCR-11-1714 [PubMed]
- 30. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005; 307:58–62. https://doi.org/10.1126/science.1104819 [PubMed]
- 31. Singhal PC, Kapasi AA, Franki N, Reddy K. Morphine-induced macrophage apoptosis: the role of transforming growth factor-beta. Immunology. 2000; 100:57–62. https://doi.org/10.1046/j.1365-2567.2000.00007.x [PubMed]
- 32. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996; 16:4604–13. https://doi.org/10.1128/MCB.16.9.4604 [PubMed]
- 33. Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997; 18:4–25. https://doi.org/10.1210/edrv.18.1.0287 [PubMed]
- 34. Hongmei Z. Extrinsic and intrinsic apoptosis signal pathway review. In Apoptosis and Medicine; IntechOpen: London, UK. 2012. https://doi.org/10.5772/50129
- 35. Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005; 4:139–63. https://doi.org/10.4161/cbt.4.2.1508 [PubMed]
- 36. Belmar J, Fesik SW. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther. 2015; 145:76–84. https://doi.org/10.1016/j.pharmthera.2014.08.003 [PubMed]
- 37. Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 1996; 87:619–28. https://doi.org/10.1016/s0092-8674(00)81382-3 [PubMed]
- 38. Wang HG, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996; 87:629–38. https://doi.org/10.1016/s0092-8674(00)81383-5 [PubMed]
- 39. Liffers ST, Munding JB, Vogt M, Kuhlmann JD, Verdoodt B, Nambiar S, Maghnouj A, Mirmohammadsadegh A, Hahn SA, Tannapfel A. MicroRNA-148a is down-regulated in human pancreatic ductal adenocarcinomas and regulates cell survival by targeting CDC25B. Lab Invest. 2011; 91:1472–9. https://doi.org/10.1038/labinvest.2011.99 [PubMed]
- 40. Fujita Y, Kojima K, Ohhashi R, Hamada N, Nozawa Y, Kitamoto A, Sato A, Kondo S, Kojima T, Deguchi T, Ito M. MiR-148a attenuates paclitaxel resistance of hormone-refractory, drug-resistant prostate cancer PC3 cells by regulating MSK1 expression. J Biol Chem. 2010; 285:19076–84. https://doi.org/10.1074/jbc.M109.079525 [PubMed]
- 41. Xu Q, Jiang Y, Yin Y, Li Q, He J, Jing Y, Qi YT, Xu Q, Li W, Lu B, Peiper SS, Jiang BH, Liu LZ. A regulatory circuit of miR-148a/152 and DNMT1 in modulating cell transformation and tumor angiogenesis through IGF-IR and IRS1. J Mol Cell Biol. 2013; 5:3–13. https://doi.org/10.1093/jmcb/mjs049 [PubMed]
- 42. Nersisyan S, Galatenko A, Chekova M, Tonevitsky A. Hypoxia-Induced miR-148a Downregulation Contributes to Poor Survival in Colorectal Cancer. Front Genet. 2021; 12:662468. https://doi.org/10.3389/fgene.2021.662468 [PubMed]
- 43. Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat Methods. 2015; 12:697. https://doi.org/10.1038/nmeth.3485 [PubMed]
- 44. Seo S, Seo K, Ki SH, Shin SM. Isorhamnetin Inhibits Reactive Oxygen Species-Dependent Hypoxia Inducible Factor (HIF)-1α Accumulation. Biol Pharm Bull. 2016; 39:1830–8. https://doi.org/10.1248/bpb.b16-00414 [PubMed]