



Introduction
The LMNA locus is complex, with variations in polyadenylation and splicing leading to the generation of all A-type lamins, which are linked to myriad nuclear structural roles and functional properties [1–3]. B-type lamins are encoded by other loci and have been attributed overlapping functions. A-type lamins have received the bulk of the attention, however, since mutations at this locus have been linked to a variety of dystrophic and progeroid syndromes [1–3]. The purpose of this review is not to provide a thorough overview of A-type lamin functions and their disease connections, but rather to evaluate one hypothesis: that altered processing of lamin A due to a reduction in Zmpste24 (aka FACE1) with age promotes aspects of the normal aging process. Several reviews have been cited herein for readers interested in broader questions around A-type lamin function and dysfunction.
Transcription of the LMNA gene produces mRNAs primarily for two proteins: prelamin A and lamin C. Prelamin A undergoes 4 post-translational steps to create mature lamin A for which the endoprotease, Zmpste24, is essential [4–7]. The prelamin A protein contains a CAAX (Cysteine-Alipathic-Alipathic-any amino acid) box at its C-terminal end (see [8, 9] for review). In humans and mice, the CAAX box of prelamin A is CSIM. The first step is farnesylation of cysteine, which is followed by cleavage of the terminal 3 amino acids by Zmpste24 or Rce1. The third step is carboxymethylation of the same cysteine. Zmpste24 is then the only known protease that can delete an additional 15 amino acids to form mature lamin A protein. If there is an imbalance with insufficient Zmpste24 to process all of the prelamin A, prelamin A accumulates in the nucleus [10].
The mutation most associated with Hutchinson-Gilford progeria syndrome is a non-coding single amino acid substitution that activates a rarely used splice site in the C-terminus of the protein. This interferes with C-terminal cleavage of lamin A by Zmpste24 resulting in a permanently farnesylated lamin A, termed progerin, which acts in a dominant fashion to promote a range of accelerated aging phenotypes comprising Hutchinson Gilford progeria syndrome (HGPS) [2, 3]. Much debate has centered on whether progerin, which is generated at low levels in absence of HGPS mutations, contributes to normal aging [11–14]. Progerin expression has been observed at very low levels in normal cells and may accumulate with age, although the latter assertion has been hard to verify [15–17].
The toxic effects of nuclear prelamin A are similar to that of progerin and resemble aspects of premature aging, but with a slower onset than progerin [18, 19]. Here, we present evidence supporting the hypothesis that declining levels of Zmpste24 with age contribute to an elevated level of prelamin A and it may be this lamin A variant that drives aspects of normal aging pathology.
Zmpste24 and accelerated aging
In both human patients and animal models, mutations leading to the expression of progerin cause a segmental progeria syndrome in which a subset of features of accelerated aging are present [11, 20]. This is also the case for mice lacking ZMPSTE24−/−, which survive about 5 months of age [6] and show both molecular and physiologic features of accelerated aging. These include genome instability [21], age-related bone loss [18], oxidative damage [22], cell senescence [19], altered epigenetic patterns [23], similarities in skeletal muscle decline [24], reduced adult stem cell function [25, 26], and altered age-related cell signaling pathways [27, 28]. Many of these phenotypes are described in more detail below (Table 1). Patients with the laminopathy, restrictive dermopathy (RD), have mutations in either ZMPSTE24 or LMNA, the latter associated with altered processing and the accumulation of prelamin A [4, 29]. RD has some phenotypes of accelerated aging; however, the condition is often very early onset and severe, making comparison with normal aging more challenging.
Table 1. Phenotypes associated with prelamin a expression that are associated with aging and/or progerin expression.
| Phenotype associated with prelamin a expression | Aging | Progerin expression | Reference(s) |
| Autophagy Defects | ✔ | ✔ | [27, 125] |
| Cellular Senescence | ✔ | ✔ | [10, 125–127] |
| DNA Damage | ✔ | ✔ | [21, 67, 125, 128] |
| Dysmorphic Nuclei | ✔ | [10, 126, 129, 130] | |
| Epigenetic Dysregulation | ✔ | ✔ | [131] |
| Heterochromatin Alterations | ✔ | ✔ | [30, 56, 110] |
Evidence for reduced expression of zmpste24 during aging – in vitro studies
Much of the current data supporting a decline in Zmpste24 protein levels during the aging process comes from analysis of cells ex vivo. Here, we summarize that data while emphasizing the need for in vivo studies in mice and human tissue samples. Figure 1 details the regulatory events that dictate Zmpste24 levels and activity during aging.
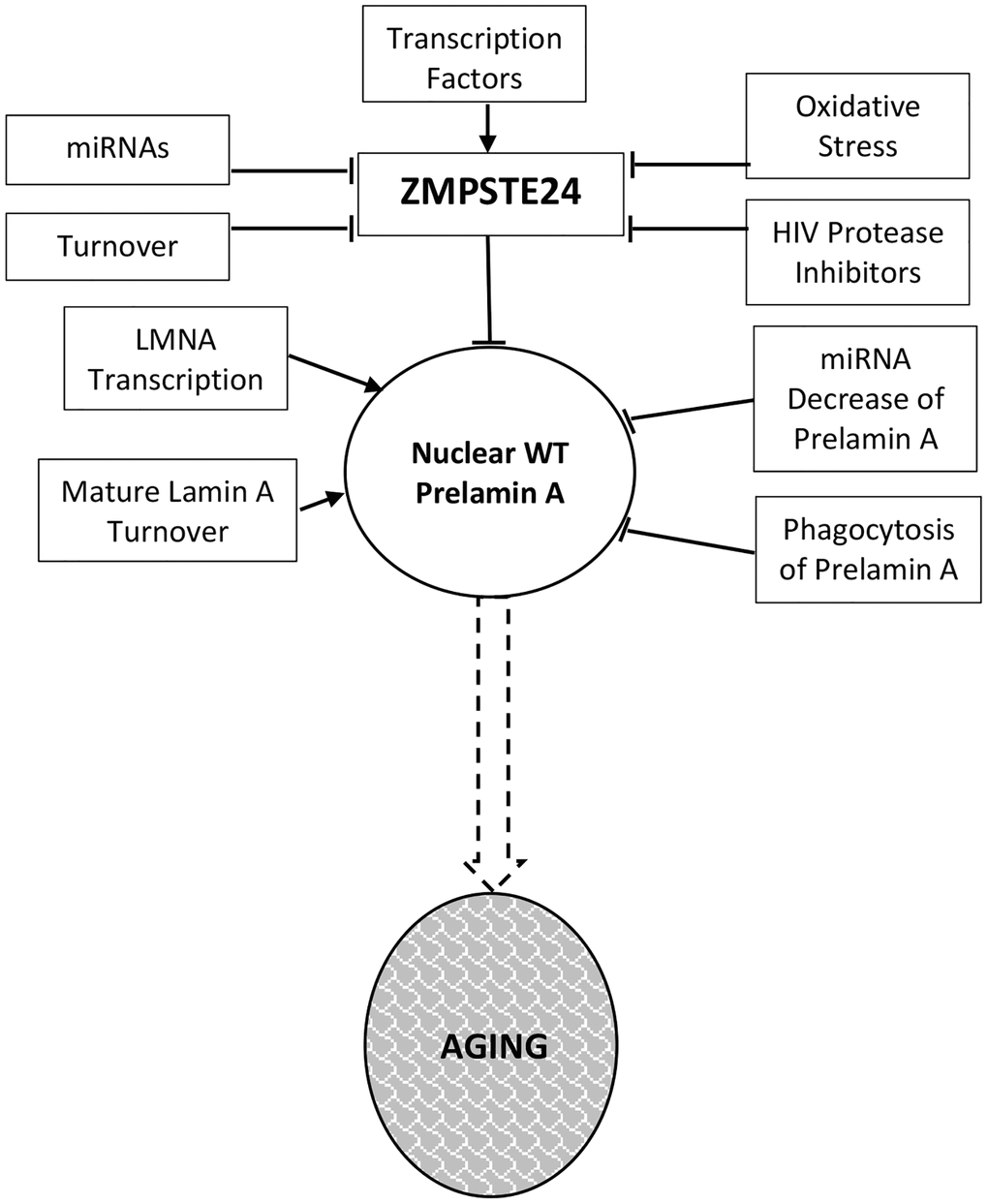
Figure 1. Prelamin a regulation in normal aging. Levels and activity of Zmpste24 are regulated in several ways related to aging. Reduced enzymatic activity is proposed to lead to increased levels of prelamin A and consequent aging phenotypes. These pathways are described throughout the text.
Skin fibroblasts of centenarians
A study of skin fibroblasts collected from young (8–35) and older (65–80) individuals, as well as centenarians (95–105) was performed and, after 6 passages in culture, the cells were assayed for aging properties [30]. Evident in cells from centenarians (over 100), but not 65–80 year old individuals, is reduced mRNA and protein levels of Zmpste24 and an accompanying increase in unprocessed lamin A. One challenge of studying centenarians is that the control group, people from the same generation who did not age well, are not around from which to collect material. Thus, when changes are observed such as those described above, two potential explanations are possible. First, it may be that ZMPSTE24 expression declines with age, implying that this is a property of normal aging. Since levels are not reduced in the 65–80 year old group, this would presumably be a late event in aging. Second, it is possible that centenarians have low levels throughout life and that this serves some protective role. We favor the former, given that other evidence indicates that Zmpste24 levels decline with age, as discussed below, but clearly more studies need to be performed.
Fetal lung fibroblasts
Human primary fibroblasts derived from fetal lung are often used for cell senescence studies. These cells will undergo senescence after prolonged serial passaging, or in response to a range of other induction methods. The level to which in vitro replicative senescence resembles aspects of normal physiologic aging remains debated [31]; however, recent studies indicate that in vivo cell senescence is a significant driver of aging in large part because of their unique secretory profiles termed the Senescence Associated Secretory Profile (SASP) [32]. One report has indicated that ZMPSTE24 (FACE-1) mRNA levels decline during cell senescence induced through serial passaging in these cells [33]. This finding is intriguing and although correlative, the decrease of Zmpste24 protein and resulting increase in prelamin A indicate cause and effect, but studies should be repeated in other primary fibroblast isolates and also when senescence is induced through other methods.
Mesenchymal stem cells
Mesenchymal stem cells (MSCs), which can be isolated from bone marrow, adipose and other tissues, are multi-potent, giving rise to cells in a range of tissues. These include bone, adipose, smooth muscle, cardiomyocytes, and other tissues that overlap significantly with those affected in laminopathies, making them interesting cells to study in this context. In addition, expression of progerin alters the differentiation properties of MSCs in vitro, impairing adipose differentiation. This is intriguing given that loss of adipose tissue is a hallmark of many laminopathies [34–36]. Also intriguing is that overexpression of wild-type lamin A conferred similar phenotypes to that of progerin, although to a lesser extent. One potential reason for this is that in the context of overexpression, the amount of lamin A produced may outpace the ability of Zmpste24 to confer processing, leading to elevated prelamin A. However, it cannot be ruled out that excess mature lamin A may disrupt nuclear functions through other mechanisms.
Enforced overexpression of prelamin A in human MSCs has also been reported to lead to elevated levels of proteins associated with osteogenesis [37]. This is surprising since ZMPSTE24−/− mice have higher levels of adipogenesis [18, 38] and osteoporosis [39]. Moreover, cells in aging bone marrow are thought to skew toward adipogenesis and away from osteogenesis [40, 41]. One clue to explain this apparent discrepancy may come from observations that older wild-type mice were found to have low levels of mature lamin A/C in osteoblasts [38], which could be an indirect consequence of loss of ZMPSTE24, at least with regard to mature lamin A. Knockdown of Lamin A/C was associated with increased adipogenesis [38]. Low levels of mature lamin A may counteract the effects of increased osteogenic factors observed in prelamin A overexpression. The MSCs are not immortal in culture and undergo replicative senescence. As with fibroblasts, senescence in these cells is associated with down-regulation of ZMPSTE24 and nuclear accumulation of prelamin A [42]. The mechanism involves upregulation of the microRNA miR-141-3p, which targets the 3′ UTR of ZMPSTE24, leading to reduced expression. Enforced expression of miR-141-3p induced senescence in cultured MSCs and injection of the microRNA in mice led to decreased liver expression of ZMPSTE24. The activity of HDAC1 and HDAC2 declines during replicative senescence and decreases the expression of ZMPSTE24 by upregulating miR-141-3p [42]. Increased prelamin A expression has also been proposed as a senescence marker to screen MSCs in vitro before clinical application [43]. These findings reinforce the studies in fibroblasts that reduced expression of ZMPSTE24 is associated with cell senescence and call for a wider analysis of the microRNA in tissues from aging animals.
A more recent paper found reduced Zmpste24 levels and prelamin A accumulation during cell senescence in subchondral bone mesenchymal stem cells [44]. Enforced expression of prelamin A in these cells accelerated senescence, which was associated with DNA damage, including at telomeres, and increased expression of inflammatory factors. Interestingly, the accelerated senescence phenotype could be suppressed by vitamin C, which also reduced inflammatory factors.
Endothelial cells
Premature senescence in primary human epithelial cells isolated from human umbilical vein or cord blood, can also be induced by elevated levels of prelamin A, this time induced by exposure of cells to protease inhibitors that inhibit activity of Zmpste24 [45], which evokes a similar phenotype in human bone marrow-derived MSCs [46]. Interestingly, this phenomenon was observed in both precursor and mature endothelial cells.
Vascular smooth muscle cells
The Shanahan lab found that nuclear accumulation of prelamin A in VSMC of arterial media with age and in vitro was correlated with the down regulation of ZMPSTE24 (See Figure 1 in [47]). These effects occurred both in vitro and in cells from old individuals processed ex vivo. The decline of ZMPSTE24 and the increase in nuclear prelamin A occurred before cellular senescence. The presenescent phase in VSMC included modification of migrational characteristics [48] and calcification due to increased levels of the osteogenic markers Runx2, ALP, and osteocalcin resulting primarily from greater DNA damage [49]. SASP factors were secreted by VSMC with higher levels of prelamin A. This finding is consistent with an early study that protease inhibitors that impair Zmpste24 function lead to premature senescence in VSMC [50].
The importance of the role of VSMC in atherosclerosis is increased by the report that VSMC may transdifferentiate into macrophage-like cells when proliferating into plaque in the intima in response to arterial damage or stress [51]. VSMC contribute with macrophages to foam cells in lesions in arteries [51, 52]. Prelamin A may have a role in these events. When endothelial cells from human umbilical vein or cord blood were treated with the HIV protease inhibitor Atazanavir, which inhibits Zmpste24, the authors observed nuclear accumulation of prelamin A leading to irregularly shaped nuclei, premature cellular senescence, and an increase in monocyte adhesion. The complexity of atherosclerosis involves many factors in addition to prelamin A in VSMC, foam cells, and endothelial cells as described in many articles [45, 53, 54]. The review of the role of lamins in atherosclerosis by Jiang and Ji provides additional useful information [55].
In vivo studies
Pilot studies
We have recently examined the levels of Zmpste24 in several tissues comparing mice of 9 and 22 months of age. While not definitive at this point, the studies strongly suggest age-associated changes in a tissue-dependent manner and call for further studies to be performed. We have included them to support the overall hypothesis and stimulate further research. Four tissues were examined: brain, liver, heart and gastrocnemius (skeletal muscle) and results are shown in Figure 2. In both brain and liver, we detect a significant decline in Zmpste24 levels, although this was not detected in heart and skeletal muscle. This finding calls for studies across a wider age range and in more tissues, and the levels of Zmpste24 should be correlated with prelamin A.
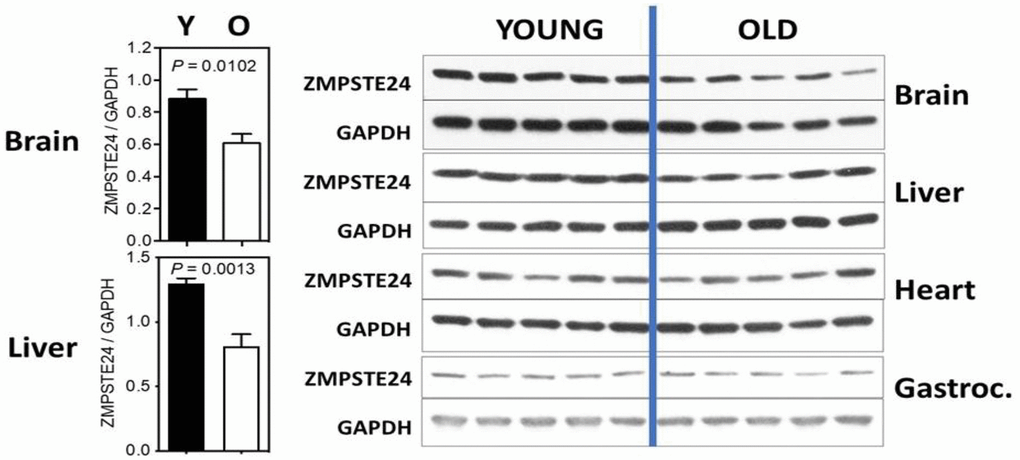
Figure 2. ZMPSTE24 protein expression during murine aging. ZMPSTE24 protein levels were determined by Western blots from brain, liver, heart, gastrocnemius, and subcutaneous fat of young (9 months of age, n = 5) and old (22 months of age, n = 5) female mice. Relative ZMPSTE24 levels (normalized to GAPDH) were quantified by densitometry using ImageJ software (http://rsb.info.nih.gov/ij/). All bars represent mean ± SEM. The statistical significance of differences between two groups (as indicated with P values) was determined using unpaired, two-tailed Student’s t-test. The antibody used was a Rabbit polyclonal to ZMPSTE24 from Abcam; Catalog number ab38450.
Aging mechanisms
Cellular senescence
Studies above indicate that declining Zmpste24 levels lead to senescent pathology in a variety of cellular contexts, raising the question of how this occurs at the mechanistic level. Several models have been proposed. Classical pathways associated with senescence involve the p53 and pRB-p16INK4A pathways. Activation of p53 and expression of p16INK4A lead to cell cycle arrest and senescence, and ablation of these pathways allow cells to escape senescence and become immortalized. p16INK4A is induced in multiple contexts associated with enhanced prelamin A expression [42, 47, 49, 56, 57], as is induction of p53 [19, 58]. Notably, induction of p16INK4A is associated with all methods to induce senescence in cell culture and, therefore, it is not surprising in the prelamin A context. This raises the question of what is happening upstream of induction of these factors.
Casein kinase 2 may be a factor linking prelamin A accumulation to cellular senescence. CK2 has long been known to be nuclear matrix-associated [59], but a recent paper shows more direct connections with lamin A, which binds to CK2 and inhibits its activity. Loss of lamin A expression leads to enhanced CK2 activity, where prelamin A accumulation is associated with its inhibition [60], in turn leading to senescence induction [60–62]. Down-regulation of CK2 is also associated with accelerated aging and oxidative stress in C. elegans [63]. Interestingly, the pro-longevity compound spermidine, which has been reported to activate CK2, was found to suppress cellular senescence in ZMPSTE24−/− MEFs and to extend the lifespan of the mice from which the cells were derived [60].
Another pathway linking prelamin A to senescence involves p62, a component of the autophagic machinery that has been linked to the aging process [64]. Enhanced autophagy, a process that involves the clearance of damaged cellular macromolecules and structures, has been implicated as a mechanism by which progerin, and more recently prelamin A, is degraded by mTOR inhibition (see below). In the context of exogenous progerin expression or reduced ZMPSTE24 expression in MSCs, DNA damage permits GATA4 to avoid p62 binding and selective degradation [65, 66]. Stabilization of the transcription factor GATA4 leads to monocyte chemoattractant protein-1 (MCP-1 expression), induction of the senescence-associated secretory phenotype and paracrine senescence [65].
DNA damage
One major candidate mechanism for the manner by which prelamin A promotes cellular senescence involves the induction of DNA damage. DNA damage, in multiple forms, or the induction of the DNA damage response have long been reported to induce cellular senescence. Several reports link expression of prelamin A to DNA damage [67], and some mechanistic studies have been conducted. For instance, cells from mice lacking ZMPSTE24 were reported early on to have elevated levels of DNA damage [21]. This phenotype has also been observed in fibroblasts from Restrictive Dermopathy patients, which have homozygous mutations in the enzyme [68]. In particular, the latter study was able to identify an increase in DNA double strand breaks [68], and both studies found defective DNA damage responses. A later study also reported increased DNA damage in smooth muscle cells expressing prelamin A [47]. Follow up studies in these cells point to a possible mechanism whereby lamin A/C is part of the DNA damage response and accumulation of prelamin A interferes with the normal role of the intermediate filament proteins [69].
DNA replication stress, in some respects a specialized form of replication stress, is not often considered. However, studies of replicative lifespan in yeast have indicated that a major driver of aging is DNA replication stress [70, 71]. Nuclear lamins have long been suspected to facilitate DNA replication [72], and recent studies have indicated that progerin or prelamin A can induce replication fork stalling, which leads to DNA breaks [73–75]. Interestingly, this leads to activation of the cGAS/STING cytosolic DNA sensing pathway and an interferon response [76]. This is perhaps consistent with increased inflammation associated with loss of ZMPSTE24 or expression of progerin, which has been previously reported [77]. Treatment of progerin expressing cells with calcitrol, an active version of vitamin D, reduces replication stress and the associated innate inflammatory response [78]. cGAS/STING signaling has been recently linked to cellular senescence and aging [79, 80], making this a pathway to explore in more detail in progeria and normal aging.
Oxidative stress
Links between oxidative stress and aging date back to the famous hypothesis by Denham Harman [81], although whether oxygen free radicals drive aging remains a matter of debate. While free radicals drive damage to a variety of cellular molecules, they also mediate critical signaling pathways, making it difficult to interpret their net effect on the aging process. Oxidative stress is also linked to a range of age-related diseases and can induce cellular senescence [82].
Increased oxidative stress is linked to reduced Zmpste24 activity [57, 83, 84], and overexpression of prelamin A in mesenchymal stem cells [85]. Interestingly, oxidative stress leads to reduced levels of Zmpste24 [47, 84, 86], possibly through upregulation of miR-141 [42]. This creates a possible feed-forward loop with oxidative damage that accumulates during aging leading to a reduction in Zmpste24 activity, which, in turn, results in more oxidative damage. More studies need to be performed in physiologic oxygen of 2 to 5% rather than in atmospheric oxygen conditions, in order to more closely resemble the in vivo environment.
Sirtuins
A-type lamins have been reported to interact with multiple Sirtuins, protein deacetylases linked to control of healthspan and lifespan [87]. With regard to SIRT1, lamin A binding leads to SIRT1 activation; however, the interaction is reduced in the presence of prelamin A, contributing to adult stem cell decline in ZMPSTE24−/− mice [88]. Lamin A also binds to and activates SIRT6, which leads to enhanced DNA repair. Again, this interaction is compromised at least in the presence of progerin (prelamin A was not reported). This may be particularly relevant as SIRT6 deficiency leads to a progeroid phenotype and overexpression of the deacetylase leads to lifespan extension [89, 90]. In culture, HGPS fibroblasts have reduced SIRT6 levels and restoration of its expression led to reduced senescence phenotypes [91]. Interestingly, SIRT6 is known to suppress LINE1 retrotransposon activation [92], which has been shown to mediate progression of cellular senescence both in SIRT6−/− and aging wild-type mice in a manner involving cGAS/STING signaling (see DNA damage section) [93, 94]. SIRT7 also represses LINE1 elements and interacts with lamin A, although whether prelamin A shows altered binding has not been reported [95].
Autophagy and mTOR signaling
The mTOR signaling pathway, of which regulation of autophagy is one major downstream pathway, is highly linked to aging [96, 97]. mTOR is a nutrient-responsive kinase that evidence indicates is aberrantly upregulated during aging. Reduced mTOR signaling, mediated genetically or with the highly specific drug rapamycin, extends lifespan in a wide range of model organisms. Evidence suggests that mTOR inhibition can also reverse aspects of aging in human studies [98, 99].
These findings make it obvious that mTOR signaling would be examined in progeria models. Initial studies in fibroblasts expressing progerin indicate that rapamycin can enhance autophagy, which is beneficial at least in part because it facilitates clearance of progerin itself [100, 101]. It was later shown to enhance cellular proliferation and reduce levels of cell senescence [102]. These studies have primarily focused on progerin, but a recent study indicates that expression of prelamin A confers similar phenotypes, and adds to previous observations by showing that the protein stimulates mTOR activation and impairs autophagy [27]. These findings suggest that more studies are needed to understand the role of mTOR in progeria and prelamin A-related normal aging.
Nucleoplasmic reticulum
When there is insufficient Zmpste24 to process prelamin A to the mature form, the nucleus may become dysmorphic [68, 103], display evaginations [104], or contain nucleoplasmic reticulum [105–107]. The term nucleoplasmic reticulum is applied to long, tubular channels that extend deep into the nucleoplasm or even pass entirely through the nucleus [107]. Some are short stubs while others are complex, branching structures. Some terminate at or near nucleoli. The nucleoplasmic reticulum is of particular interest here because it is formed during interphase by excess nuclear prelamin A, as demonstrated when Interphase prelamin A was experimentally produced by suppressing ZMPSTE24, either by siRNA or by application of an HIV protease inhibitor (PI) such as saquinavir [105, 106]. When saquinavir was removed from the media, processing resulted in mature lamin A and the number of nucleoplasmic reticulum invaginations was markedly reduced [106]. The role of nucleoplasmic reticulum formation in aging remains poorly understood and more studies are needed in aging organisms.
Stepping back, there are also numerous studies linking progerin, and to a lesser extent prelamin A, expression to loss of heterochromatin. Links to progerin are thoroughly described in a recent review [108]. Regarding prelamin A, early studies linked prelamin A to loss of chromatin organization [109, 110]. More recently, studies of lamina-associated domains (LADs) in Zmpste24−/− mice indicate altered associations with transcription factors, including Foxa2 that are similar to those of old wild-type mice [111]. More studies are needed, but understanding the heterochromatin alterations associated with altered lamin A function remains a vital area of research.
Other functions of zmpste24
Given that loss-of-function mutations in ZMPSTE24 give rise to phenotypes resembling those associated with LMNA mutations, many have assumed that the role of Zmpste24 is restricted to modifying processing of Lamin A; yet this may be too simple as other functions of Zmpste24 are known. For instance, other substrates of Zmpste24 have been identified, including proteins that are not prenylated, although the significance of these events remain largely unknown [112]. Notably, Zmpste24 assists in helping translocon pores in the endoplasmic reticulum function smoothly [113, 114]. Translocons can become clogged when proteins enter but fail to properly transverse the pore. Under these conditions, Zmpste24 cleaves clogged proteins into peptide fragments for clearance. This function may have roles in aging, where protein misfolding is increased. A recent study, however, found that ZMPSTE24 disease mutations all affected lamin A processing but only some mutants interfered with the ability of the enzyme to clear clogged proteins from the translocon [115].
Independently of its enzymatic functions, Zmpste24 also interacts with the interferon-induced transmembrane protein (IFITM) family and facilitates the role of these proteins in blocking entry of enveloped RNA and DNA viruses [116, 117]. Zmpste24 appears to impair entry of a range of viruses, including influenza A, Zika and COVID-19 [118, 119]. As a result, mice lacking ZMPSTE24 have increased viral loads and show sensitivity to influenza infection. Whether reduced Zmpste24 levels with age contribute to the increased sensitively of older individuals to viruses remains to be determined.
Questions to be addressed
We propose the following: (1) A-type nuclear lamins are involved in normal aging as well as progeria and (2) it is prelamin A resulting from declining levels of Zmpste24 that drives aspects of normal aging more than expression of progerin. We have described a variety of supporting evidence; however, comprehensive studies remain to be performed to validate this approach. It is critical to address this question given the dramatic increase in the aging population worldwide and the accompanying chronic diseases for which aging is the biggest risk factor. If the lamin A processing pathway can be validated as a driver of normal aging, a variety of new therapeutic approaches will be feasible to extend healthspan. Moreover, this may explain the longevity benefits associated with known interventions, including mTOR inhibitors and Sirtuin activators, among others.
To validate this theory, more comprehensive studies are needed to confirm that Zmpste24 levels and activity decline with aging in animal models and humans and that this is associated with elevated prelamin A levels. These studies need to be performed under optimal conditions (for instance physiological oxygen levels for cell culture) and with the best possible reagents without which interpretation of results is more complicated. Antibodies specific for prelamin A include 3C8 [120] and PL-1C7 [9]. Antibodies specific for Zmpste24 include PA1-16965 [113], 205-8C10 (Daiichi Chemical), and ab38450 (Abcam). We also note that many publications show prelamin A levels without accompanying levels of lamin A and/or without detection of a slower migrating prelamin A band. This precludes determination of ratios of different lamin A isoforms. It may be the ratio of prelamin A to mature A-type lamins that best define associated phenotypes and this should be measured in research studies, if at all possible.
In mice, Zmpste24 and prelamin A levels should be carefully examined in multiple tissues of mice at a variety of ages. Ideally, a variety of associated factors should be analyzed, including levels of (1) other lamin isoforms, (2) microRNAs associated with A-type lamin and ZMPSTE24 expression, (3) senescence factors and (4) markers of the activity of related pathways such as mTOR. If, as expected, declines in Zmpste24 levels are observed, it may be worth engineering mice lacking the 3′UTR sites for miR-141-3p and miR-335 binding in ZMPSTE24 [42]. Another genetically modified mouse model of interest would be engineered to overexpress ZMPSTE24 systemically or in a tissue-specific manner. The prediction would be that these mice would have improved healthspan and lifespan.
Human studies are also needed, including further analysis of cells isolated from humans at different age ranges for the same parameters as those described for murine studies. In addition, muscle or skin biopsies should be tested from different age ranges. Muscle may be of particular importance since mTOR signaling is known to increase in this tissue with age [121]. Finally, human longevity intervention studies should consider examining the levels of prelamin A and Zmpste24 whenever possible. Interventions could include microRNA therapeutics, for instance mimicking the effects of miR-9 to reduce LMNA expression [122, 123] or inhibiting miR-141-3p through antimiRs [42, 124], strategies to enhance degradation of prelamin A, for instance by driving autophagic clearance [101], or altering transcription levels of ZMPSTE24. The appropriate strategy will clearly await a better mechanistic understanding regarding the reasons Zmpste24 levels decline with age.
Understanding the pathways that modulate the aging process is critical toward developing strategies to extend human healthspan, and to assist individuals with progeroid disorders. It has long been debated to what extent the mechanisms of aging and progeria overlap. Loss of Zmpste24 may be a connecting feature and, if correct, serve as a target for present and future longevity interventions.
Author Contributions
S. R. Primmer conceived the hypothesis, reviewed the literature, and wrote the initial draft; Chen-Yu Liao and Oona M.P. Kummert performed the Pilot Study with preliminary analyses of murine tissues; B. K. Kennedy participated in forming the hypothesis, and edited and rewrote the final copy.
Acknowledgments
The authors would like to thank Amanda M. Bair and Delana M. Miller for assistance with preliminary analyses of murine tissues.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement
Murine studies in this manuscript were approved by an Institutional Animal Care and Use Committee (IACUC) at the Buck Institute for Research on Aging.
Funding
Support of this study was provided by NUHSRO/2020/114.
References
- 1. Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell. 2013; 152:1365–75. https://doi.org/10.1016/j.cell.2013.02.015 [PubMed]
- 2. Foo MXR, Ong PF, Dreesen O. Premature aging syndromes: From patients to mechanism. J Dermatol Sci. 2019; 96:58–65. https://doi.org/10.1016/j.jdermsci.2019.10.003 [PubMed]
- 3. Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol. 2016; 145:401–17. https://doi.org/10.1007/s00418-016-1411-1 [PubMed]
- 4. Navarro CL, Cadiñanos J, De Sandre-Giovannoli A, Bernard R, Courrier S, Boccaccio I, Boyer A, Kleijer WJ, Wagner A, Giuliano F, Beemer FA, Freije JM, Cau P, et al. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet. 2005; 14:1503–13. https://doi.org/10.1093/hmg/ddi159 [PubMed]
- 5. Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002; 99:13049–54. https://doi.org/10.1073/pnas.192460799 [PubMed]
- 6. Pendás AM, Zhou Z, Cadiñanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodríguez F, Tryggvason K, López-Otín C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002; 31:94–9. https://doi.org/10.1038/ng871 [PubMed]
- 7. Corrigan DP, Kuszczak D, Rusinol AE, Thewke DP, Hrycyna CA, Michaelis S, Sinensky MS. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J. 2005; 387:129–38. https://doi.org/10.1042/BJ20041359 [PubMed]
- 8. Davies BS, Fong LG, Yang SH, Coffinier C, Young SG. The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet. 2009; 10:153–74. https://doi.org/10.1146/annurev-genom-082908-150150 [PubMed]
- 9. Casasola A, Scalzo D, Nandakumar V, Halow J, Recillas-Targa F, Groudine M, Rincón-Arano H. Prelamin A processing, accumulation and distribution in normal cells and laminopathy disorders. Nucleus. 2016; 7:84–102. https://doi.org/10.1080/19491034.2016.1150397 [PubMed]
- 10. Candelario J, Sudhakar S, Navarro S, Reddy S, Comai L. Perturbation of wild-type lamin A metabolism results in a progeroid phenotype. Aging Cell. 2008; 7:355–67. https://doi.org/10.1111/j.1474-9726.2008.00393.x [PubMed]
- 11. Ashapkin VV, Kutueva LI, Kurchashova SY, Kireev II. Are There Common Mechanisms Between the Hutchinson-Gilford Progeria Syndrome and Natural Aging? Front Genet. 2019; 10:455. https://doi.org/10.3389/fgene.2019.00455 [PubMed]
- 12. Reddy S, Comai L. Recent advances in understanding the role of lamins in health and disease. F1000Res. 2016; 5:2536. https://doi.org/10.12688/f1000research.9260.1 [PubMed]
- 13. Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 2010; 11:567–78. https://doi.org/10.1038/nrm2944 [PubMed]
- 14. Shimi T, Goldman RD. Nuclear lamins and oxidative stress in cell proliferation and longevity. Adv Exp Med Biol. 2014; 773:415–30. https://doi.org/10.1007/978-1-4899-8032-8_19 [PubMed]
- 15. Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A. 2007; 104:4949–54. https://doi.org/10.1073/pnas.0611640104 [PubMed]
- 16. McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One. 2007; 2:e1269. https://doi.org/10.1371/journal.pone.0001269 [PubMed]
- 17. Luo YB, Mitrpant C, Johnsen RD, Fabian VA, Fletcher S, Mastaglia FL, Wilton SD. Investigation of age-related changes in LMNA splicing and expression of progerin in human skeletal muscles. Int J Clin Exp Pathol. 2013; 6:2778–86. [PubMed]
- 18. Rivas D, Li W, Akter R, Henderson JE, Duque G. Accelerated features of age-related bone loss in zmpste24 metalloproteinase-deficient mice. J Gerontol A Biol Sci Med Sci. 2009; 64:1015–24. https://doi.org/10.1093/gerona/glp089 [PubMed]
- 19. Varela I, Cadiñanos J, Pendás AM, Gutiérrez-Fernández A, Folgueras AR, Sánchez LM, Zhou Z, Rodríguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, López-Otín C. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005; 437:564–8. https://doi.org/10.1038/nature04019 [PubMed]
- 20. Cenni V, Capanni C, Mattioli E, Schena E, Squarzoni S, Bacalini MG, Garagnani P, Salvioli S, Franceschi C, Lattanzi G. Lamin A involvement in ageing processes. Ageing Res Rev. 2020; 62:101073. https://doi.org/10.1016/j.arr.2020.101073 [PubMed]
- 21. Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadiñanos J, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005; 11:780–5. https://doi.org/10.1038/nm1266 [PubMed]
- 22. Peinado JR, Quirós PM, Pulido MR, Mariño G, Martínez-Chantar ML, Vázquez-Martínez R, Freije JM, López-Otín C, Malagón MM. Proteomic profiling of adipose tissue from Zmpste24-/- mice, a model of lipodystrophy and premature aging, reveals major changes in mitochondrial function and vimentin processing. Mol Cell Proteomics. 2011; 10:1–17. https://doi.org/10.1074/mcp.M111.008094 [PubMed]
- 23. Osorio FG, Varela I, Lara E, Puente XS, Espada J, Santoro R, Freije JM, Fraga MF, López-Otín C. Nuclear envelope alterations generate an aging-like epigenetic pattern in mice deficient in Zmpste24 metalloprotease. Aging Cell. 2010; 9:947–57. https://doi.org/10.1111/j.1474-9726.2010.00621.x [PubMed]
- 24. Greising SM, Call JA, Lund TC, Blazar BR, Tolar J, Lowe DA. Skeletal muscle contractile function and neuromuscular performance in Zmpste24 -/- mice, a murine model of human progeria. Age (Dordr). 2012; 34:805–19. https://doi.org/10.1007/s11357-011-9281-x [PubMed]
- 25. Song M, Lavasani M, Thompson SD, Lu A, Ahani B, Huard J. Muscle-derived stem/progenitor cell dysfunction in Zmpste24-deficient progeroid mice limits muscle regeneration. Stem Cell Res Ther. 2013; 4:33. https://doi.org/10.1186/scrt183 [PubMed]
- 26. Espada J, Varela I, Flores I, Ugalde AP, Cadiñanos J, Pendás AM, Stewart CL, Tryggvason K, Blasco MA, Freije JM, López-Otín C. Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J Cell Biol. 2008; 181:27–35. https://doi.org/10.1083/jcb.200801096 [PubMed]
- 27. Pan X, Jiang B, Wu X, Xu H, Cao S, Bai N, Li X, Yi F, Guo Q, Guo W, Song X, Meng F, Li X, et al. Accumulation of prelamin A induces premature aging through mTOR overactivation. FASEB J. 2020; 34:7905–14. https://doi.org/10.1096/fj.201903048RR [PubMed]
- 28. Mariño G, Ugalde AP, Fernández AF, Osorio FG, Fueyo A, Freije JM, López-Otín C. Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proc Natl Acad Sci U S A. 2010; 107:16268–73. https://doi.org/10.1073/pnas.1002696107 [PubMed]
- 29. Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Geneviève D, Hadj-Rabia S, Gaudy-Marqueste C, Smitt HS, Vabres P, Faivre L, Verloes A, Van Essen T, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004; 13:2493–503. https://doi.org/10.1093/hmg/ddh265 [PubMed]
- 30. Lattanzi G, Ortolani M, Columbaro M, Prencipe S, Mattioli E, Lanzarini C, Maraldi NM, Cenni V, Garagnani P, Salvioli S, Storci G, Bonafè M, Capanni C, Franceschi C. Lamins are rapamycin targets that impact human longevity: a study in centenarians. J Cell Sci. 2014; 127:147–57. https://doi.org/10.1242/jcs.133983 [PubMed]
- 31. de Magalhães JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev. 2018; 170:2–9. https://doi.org/10.1016/j.mad.2017.07.001 [PubMed]
- 32. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018; 128:1238–46. https://doi.org/10.1172/JCI95148 [PubMed]
- 33. Ukekawa R, Miki K, Fujii M, Hirano H, Ayusawa D. Accumulation of multiple forms of lamin A with down-regulation of FACE-1 suppresses growth in senescent human cells. Genes Cells. 2007; 12:397–406. https://doi.org/10.1111/j.1365-2443.2007.01057.x [PubMed]
- 34. Capeau J, Magré J, Lascols O, Caron M, Béréziat V, Vigouroux C, Bastard JP. Diseases of adipose tissue: genetic and acquired lipodystrophies. Biochem Soc Trans. 2005; 33:1073–7. https://doi.org/10.1042/BST0331073 [PubMed]
- 35. Decostre V, Ben Yaou R, Bonne G. Laminopathies affecting skeletal and cardiac muscles: clinical and pathophysiological aspects. Acta Myol. 2005; 24:104–9. [PubMed]
- 36. Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, Schmidt H, Brabant G, Kumar S, Durrington PN, Gregory S, O'Rahilly S, Trembath RC. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000; 24:153–6. https://doi.org/10.1038/72807 [PubMed]
- 37. Infante A, Rodríguez CI. Secretome analysis of in vitro aged human mesenchymal stem cells reveals IGFBP7 as a putative factor for promoting osteogenesis. Sci Rep. 2018; 8:4632. https://doi.org/10.1038/s41598-018-22855-z [PubMed]
- 38. Vidal C, Bermeo S, Fatkin D, Duque G. Role of the nuclear envelope in the pathogenesis of age-related bone loss and osteoporosis. Bonekey Rep. 2012; 1:62. https://doi.org/10.1038/bonekey.2012.62 [PubMed]
- 39. Fei D, Zhang Y, Wu J, Zhang H, Liu A, He X, Wang J, Li B, Wang Q, Jin Y. Cav 1.2 regulates osteogenesis of bone marrow-derived mesenchymal stem cells via canonical Wnt pathway in age-related osteoporosis. Aging Cell. 2019; 18:e12967. https://doi.org/10.1111/acel.12967 [PubMed]
- 40. Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology. 2001; 2:165–71. https://doi.org/10.1023/a:1011513223894 [PubMed]
- 41. Kajkenova O, Lecka-Czernik B, Gubrij I, Hauser SP, Takahashi K, Parfitt AM, Jilka RL, Manolagas SC, Lipschitz DA. Increased adipogenesis and myelopoiesis in the bone marrow of SAMP6, a murine model of defective osteoblastogenesis and low turnover osteopenia. J Bone Miner Res. 1997; 12:1772–9. https://doi.org/10.1359/jbmr.1997.12.11.1772 [PubMed]
- 42. Yu KR, Lee S, Jung JW, Hong IS, Kim HS, Seo Y, Shin TH, Kang KS. MicroRNA-141-3p plays a role in human mesenchymal stem cell aging by directly targeting ZMPSTE24. J Cell Sci. 2013; 126:5422–31. https://doi.org/10.1242/jcs.133314 [PubMed]
- 43. Bellotti C, Capanni C, Lattanzi G, Donati D, Lucarelli E, Duchi S. Detection of mesenchymal stem cells senescence by prelamin A accumulation at the nuclear level. Springerplus. 2016; 5:1427. https://doi.org/10.1186/s40064-016-3091-7 [PubMed]
- 44. Qu YN, Zhang L, Wang T, Zhang HY, Yang ZJ, Yuan FF, Wang Y, Li SW, Jiang XX, Xie XH. Vitamin C Treatment Rescues Prelamin A-Induced Premature Senescence of Subchondral Bone Mesenchymal Stem Cells. Stem Cells Int. 2020; 2020:3150716. https://doi.org/10.1155/2020/3150716 [PubMed]
- 45. Bonello-Palot N, Simoncini S, Robert S, Bourgeois P, Sabatier F, Levy N, Dignat-George F, Badens C. Prelamin A accumulation in endothelial cells induces premature senescence and functional impairment. Atherosclerosis. 2014; 237:45–52. https://doi.org/10.1016/j.atherosclerosis.2014.08.036 [PubMed]
- 46. Hernandez-Vallejo SJ, Beaupere C, Larghero J, Capeau J, Lagathu C. HIV protease inhibitors induce senescence and alter osteoblastic potential of human bone marrow mesenchymal stem cells: beneficial effect of pravastatin. Aging Cell. 2013; 12:955–65. https://doi.org/10.1111/acel.12119 [PubMed]
- 47. Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, Shroff R, Skepper J, Shanahan CM. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. 2010; 121:2200–10. https://doi.org/10.1161/CIRCULATIONAHA.109.902056 [PubMed]
- 48. Porter LJ, Holt MR, Soong D, Shanahan CM, Warren DT. Prelamin A Accumulation Attenuates Rac1 Activity and Increases the Intrinsic Migrational Persistence of Aged Vascular Smooth Muscle Cells. Cells. 2016; 5:41. https://doi.org/10.3390/cells5040041 [PubMed]
- 49. Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. 2013; 112:e99–109. https://doi.org/10.1161/CIRCRESAHA.111.300543 [PubMed]
- 50. Lefèvre C, Auclair M, Boccara F, Bastard JP, Capeau J, Vigouroux C, Caron-Debarle M. Premature senescence of vascular cells is induced by HIV protease inhibitors: implication of prelamin A and reversion by statin. Arterioscler Thromb Vasc Biol. 2010; 30:2611–20. https://doi.org/10.1161/ATVBAHA.110.213603 [PubMed]
- 51. Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, Feil R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014; 115:662–7. https://doi.org/10.1161/CIRCRESAHA.115.304634 [PubMed]
- 52. Allahverdian S, Pannu PS, Francis GA. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovasc Res. 2012; 95:165–72. https://doi.org/10.1093/cvr/cvs094 [PubMed]
- 53. Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res. 2006; 69:614–24. https://doi.org/10.1016/j.cardiores.2005.08.002 [PubMed]
- 54. Newby AC, Zaltsman AB. Fibrous cap formation or destruction--the critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc Res. 1999; 41:345–60. [PubMed]
- 55. Jiang Y, Ji JY. Understanding lamin proteins and their roles in aging and cardiovascular diseases. Life Sci. 2018; 212:20–9. https://doi.org/10.1016/j.lfs.2018.09.026 [PubMed]
- 56. Liu J, Yin X, Liu B, Zheng H, Zhou G, Gong L, Li M, Li X, Wang Y, Hu J, Krishnan V, Zhou Z, Wang Z. HP1α mediates defective heterochromatin repair and accelerates senescence in Zmpste24-deficient cells. Cell Cycle. 2014; 13:1237–47. https://doi.org/10.4161/cc.28105 [PubMed]
- 57. Caron M, Auclair M, Donadille B, Béréziat V, Guerci B, Laville M, Narbonne H, Bodemer C, Lascols O, Capeau J, Vigouroux C. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007; 14:1759–67. https://doi.org/10.1038/sj.cdd.4402197 [PubMed]
- 58. Osorio FG, Obaya AJ, López-Otín C, Freije JM. Accelerated ageing: from mechanism to therapy through animal models. Transgenic Res. 2009; 18:7–15. https://doi.org/10.1007/s11248-008-9226-z [PubMed]
- 59. Li H, Roux SJ. Casein kinase II protein kinase is bound to lamina-matrix and phosphorylates lamin-like protein in isolated pea nuclei. Proc Natl Acad Sci U S A. 1992; 89:8434–8. https://doi.org/10.1073/pnas.89.18.8434 [PubMed]
- 60. Ao Y, Zhang J, Liu Z, Qian M, Li Y, Wu Z, Sun P, Wu J, Bei W, Wen J, Wu X, Li F, Zhou Z, et al. Lamin A buffers CK2 kinase activity to modulate aging in a progeria mouse model. Sci Adv. 2019; 5:eaav5078. https://doi.org/10.1126/sciadv.aav5078 [PubMed]
- 61. Chang BD, Watanabe K, Broude EV, Fang J, Poole JC, Kalinichenko TV, Roninson IB. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: implications for carcinogenesis, senescence, and age-related diseases. Proc Natl Acad Sci U S A. 2000; 97:4291–6. https://doi.org/10.1073/pnas.97.8.4291 [PubMed]
- 62. Ryu SW, Woo JH, Kim YH, Lee YS, Park JW, Bae YS. Downregulation of protein kinase CKII is associated with cellular senescence. FEBS Lett. 2006; 580:988–94. https://doi.org/10.1016/j.febslet.2006.01.028 [PubMed]
- 63. Park JH, Lee JH, Park JW, Kim DY, Hahm JH, Nam HG, Bae YS. Downregulation of protein kinase CK2 activity induces age-related biomarkers in C. elegans. Oncotarget. 2017; 8:36950–63. https://doi.org/10.18632/oncotarget.16939 [PubMed]
- 64. Fan X, Huang T, Tong Y, Fan Z, Yang Z, Yang D, Mao X, Yang M. p62 works as a hub modulation in the ageing process. Ageing Res Rev. 2022; 73:101538. https://doi.org/10.1016/j.arr.2021.101538 [PubMed]
- 65. Lee JY, Yu KR, Lee BC, Kang I, Kim JJ, Jung EJ, Kim HS, Seo Y, Choi SW, Kang KS. GATA4-dependent regulation of the secretory phenotype via MCP-1 underlies lamin A-mediated human mesenchymal stem cell aging. Exp Mol Med. 2018; 50:1–12. https://doi.org/10.1038/s12276-018-0092-3 [PubMed]
- 66. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015; 349:aaa5612. https://doi.org/10.1126/science.aaa5612 [PubMed]
- 67. Musich PR, Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany NY). 2009; 1:28–37. https://doi.org/10.18632/aging.100012 [PubMed]
- 68. Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci. 2006; 119:4644–9. https://doi.org/10.1242/jcs.03263 [PubMed]
- 69. Warren DT, Tajsic T, Porter LJ, Minaisah RM, Cobb A, Jacob A, Rajgor D, Zhang QP, Shanahan CM. Nesprin-2-dependent ERK1/2 compartmentalisation regulates the DNA damage response in vascular smooth muscle cell ageing. Cell Death Differ. 2015; 22:1540–50. https://doi.org/10.1038/cdd.2015.12 [PubMed]
- 70. Kwan EX, Foss EJ, Tsuchiyama S, Alvino GM, Kruglyak L, Kaeberlein M, Raghuraman MK, Brewer BJ, Kennedy BK, Bedalov A. A natural polymorphism in rDNA replication origins links origin activation with calorie restriction and lifespan. PLoS Genet. 2013; 9:e1003329. https://doi.org/10.1371/journal.pgen.1003329 [PubMed]
- 71. Green LA, Yates JF. Influence of pseudodiagnostic information on the evaluation of ischemic heart disease. Ann Emerg Med. 1995; 25:451–7. https://doi.org/10.1016/s0196-0644(95)70257-1 [PubMed]
- 72. Graziano S, Kreienkamp R, Coll-Bonfill N, Gonzalo S. Causes and consequences of genomic instability in laminopathies: Replication stress and interferon response. Nucleus. 2018; 9:258–75. https://doi.org/10.1080/19491034.2018.1454168 [PubMed]
- 73. Cobb AM, Murray TV, Warren DT, Liu Y, Shanahan CM. Disruption of PCNA-lamins A/C interactions by prelamin A induces DNA replication fork stalling. Nucleus. 2016; 7:498–511. https://doi.org/10.1080/19491034.2016.1239685 [PubMed]
- 74. Chojnowski A, Ong PF, Foo MXR, Liebl D, Hor LP, Stewart CL, Dreesen O. Heterochromatin loss as a determinant of progerin-induced DNA damage in Hutchinson-Gilford Progeria. Aging Cell. 2020; 19:e13108. https://doi.org/10.1111/acel.13108 [PubMed]
- 75. Hilton BA, Liu J, Cartwright BM, Liu Y, Breitman M, Wang Y, Jones R, Tang H, Rusinol A, Musich PR, Zou Y. Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. FASEB J. 2017; 31:3882–93. https://doi.org/10.1096/fj.201700014R [PubMed]
- 76. Kreienkamp R, Graziano S, Coll-Bonfill N, Bedia-Diaz G, Cybulla E, Vindigni A, Dorsett D, Kubben N, Batista LFZ, Gonzalo S. A Cell-Intrinsic Interferon-like Response Links Replication Stress to Cellular Aging Caused by Progerin. Cell Rep. 2018; 22:2006–15. https://doi.org/10.1016/j.celrep.2018.01.090 [PubMed]
- 77. Osorio FG, Bárcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM, López-Otín C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012; 26:2311–24. https://doi.org/10.1101/gad.197954.112 [PubMed]
- 78. Coll-Bonfill N, Cancado de Faria R, Bhoopatiraju S, Gonzalo S. Calcitriol Prevents RAD51 Loss and cGAS-STING-IFN Response Triggered by Progerin. Proteomics. 2020; 20:e1800406. https://doi.org/10.1002/pmic.201800406 [PubMed]
- 79. Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017; 19:1061–70. https://doi.org/10.1038/ncb3586 [PubMed]
- 80. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, Capell BC, Xu C, Xu M, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017; 550:402–6. https://doi.org/10.1038/nature24050 [PubMed]
- 81. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956; 11:298–300. https://doi.org/10.1093/geronj/11.3.298 [PubMed]
- 82. Dodig S, Čepelak I, Pavić I. Hallmarks of senescence and aging. Biochem Med (Zagreb). 2019; 29:030501. https://doi.org/10.11613/BM.2019.030501 [PubMed]
- 83. Sieprath T, Corne TD, Nooteboom M, Grootaert C, Rajkovic A, Buysschaert B, Robijns J, Broers JL, Ramaekers FC, Koopman WJ, Willems PH, De Vos WH. Sustained accumulation of prelamin A and depletion of lamin A/C both cause oxidative stress and mitochondrial dysfunction but induce different cell fates. Nucleus. 2015; 6:236–46. https://doi.org/10.1080/19491034.2015.1050568 [PubMed]
- 84. Muteliefu G, Shimizu H, Enomoto A, Nishijima F, Takahashi M, Niwa T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am J Physiol Cell Physiol. 2012; 303:C126–34. https://doi.org/10.1152/ajpcell.00329.2011 [PubMed]
- 85. Mateos J, De la Fuente A, Lesende-Rodriguez I, Fernández-Pernas P, Arufe MC, Blanco FJ. Lamin A deregulation in human mesenchymal stem cells promotes an impairment in their chondrogenic potential and imbalance in their response to oxidative stress. Stem Cell Res. 2013; 11:1137–48. https://doi.org/10.1016/j.scr.2013.07.004 [PubMed]
- 86. Afonso P, Auclair M, Boccara F, Vantyghem MC, Katlama C, Capeau J, Vigouroux C, Caron-Debarle M. LMNA mutations resulting in lipodystrophy and HIV protease inhibitors trigger vascular smooth muscle cell senescence and calcification: Role of ZMPSTE24 downregulation. Atherosclerosis. 2016; 245:200–11. https://doi.org/10.1016/j.atherosclerosis.2015.12.012 [PubMed]
- 87. Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol. 2016; 17:679–90. https://doi.org/10.1038/nrm.2016.93 [PubMed]
- 88. Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, Kaeberlein M, Tryggvason K, Zhou Z. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 2012; 16:738–50. https://doi.org/10.1016/j.cmet.2012.11.007 [PubMed]
- 89. Roichman A, Elhanati S, Aon MA, Abramovich I, Di Francesco A, Shahar Y, Avivi MY, Shurgi M, Rubinstein A, Wiesner Y, Shuchami A, Petrover Z, Lebenthal-Loinger I, et al. Restoration of energy homeostasis by SIRT6 extends healthy lifespan. Nat Commun. 2021; 12:3208. https://doi.org/10.1038/s41467-021-23545-7 [PubMed]
- 90. Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012; 483:218–21. https://doi.org/10.1038/nature10815 [PubMed]
- 91. Endisha H, Merrill-Schools J, Zhao M, Bristol M, Wang X, Kubben N, Elmore LW. Restoring SIRT6 Expression in Hutchinson-Gilford Progeria Syndrome Cells Impedes Premature Senescence and Formation of Dysmorphic Nuclei. Pathobiology. 2015; 82:9–20. https://doi.org/10.1159/000368856 [PubMed]
- 92. Van Meter M, Kashyap M, Rezazadeh S, Geneva AJ, Morello TD, Seluanov A, Gorbunova V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat Commun. 2014; 5:5011. https://doi.org/10.1038/ncomms6011 [PubMed]
- 93. Simon M, Van Meter M, Ablaeva J, Ke Z, Gonzalez RS, Taguchi T, De Cecco M, Leonova KI, Kogan V, Helfand SL, Neretti N, Roichman A, Cohen HY, et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab. 2019; 29:871–85.e5. https://doi.org/10.1016/j.cmet.2019.02.014 [PubMed]
- 94. De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, Le O, Beauséjour C, Ambati J, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019; 566:73–8. https://doi.org/10.1038/s41586-018-0784-9 [PubMed]
- 95. Vazquez BN, Thackray JK, Simonet NG, Chahar S, Kane-Goldsmith N, Newkirk SJ, Lee S, Xing J, Verzi MP, An W, Vaquero A, Tischfield JA, Serrano L. SIRT7 mediates L1 elements transcriptional repression and their association with the nuclear lamina. Nucleic Acids Res. 2019; 47:7870–85. https://doi.org/10.1093/nar/gkz519 [PubMed]
- 96. Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013; 493:338–45. https://doi.org/10.1038/nature11861 [PubMed]
- 97. Kennedy BK, Lamming DW. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016; 23:990–1003. https://doi.org/10.1016/j.cmet.2016.05.009 [PubMed]
- 98. Mannick JB, Teo G, Bernardo P, Quinn D, Russell K, Klickstein L, Marshall W, Shergill S. Targeting the biology of ageing with mTOR inhibitors to improve immune function in older adults: phase 2b and phase 3 randomised trials. Lancet Healthy Longev. 2021; 2:e250–62. https://doi.org/10.1016/S2666-7568(21)00062-3 [PubMed]
- 99. Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, Watkins M, Shavlakadze T, Zhou W, Quinn D, Glass DJ, Klickstein LB. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med. 2018; 10:eaaq1564. https://doi.org/10.1126/scitranslmed.aaq1564 [PubMed]
- 100. Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011; 3:89ra58. https://doi.org/10.1126/scitranslmed.3002346 [PubMed]
- 101. Cenni V, Capanni C, Columbaro M, Ortolani M, D'Apice MR, Novelli G, Fini M, Marmiroli S, Scarano E, Maraldi NM, Squarzoni S, Prencipe S, Lattanzi G. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur J Histochem. 2011; 55:e36. https://doi.org/10.4081/ejh.2011.e36 [PubMed]
- 102. DuBose AJ, Lichtenstein ST, Petrash NM, Erdos MR, Gordon LB, Collins FS. Everolimus rescues multiple cellular defects in laminopathy-patient fibroblasts. Proc Natl Acad Sci U S A. 2018; 115:4206–11. https://doi.org/10.1073/pnas.1802811115 [PubMed]
- 103. Caron M, Auclair M, Sterlingot H, Kornprobst M, Capeau J. Some HIV protease inhibitors alter lamin A/C maturation and stability, SREBP-1 nuclear localization and adipocyte differentiation. AIDS. 2003; 17:2437–44. https://doi.org/10.1097/00002030-200311210-00005 [PubMed]
- 104. Lu X, Djabali K. Autophagic Removal of Farnesylated Carboxy-Terminal Lamin Peptides. Cells. 2018; 7:33. https://doi.org/10.3390/cells7040033 [PubMed]
- 105. Goulbourne CN, Malhas AN, Vaux DJ. The induction of a nucleoplasmic reticulum by prelamin A accumulation requires CTP:phosphocholine cytidylyltransferase-α. J Cell Sci. 2011; 124:4253–66. https://doi.org/10.1242/jcs.091009 [PubMed]
- 106. Drozdz MM, Jiang H, Pytowski L, Grovenor C, Vaux DJ. Formation of a nucleoplasmic reticulum requires de novo assembly of nascent phospholipids and shows preferential incorporation of nascent lamins. Sci Rep. 2017; 7:7454. https://doi.org/10.1038/s41598-017-07614-w [PubMed]
- 107. Fricker M, Hollinshead M, White N, Vaux D. Interphase nuclei of many mammalian cell types contain deep, dynamic, tubular membrane-bound invaginations of the nuclear envelope. J Cell Biol. 1997; 136:531–44. https://doi.org/10.1083/jcb.136.3.531 [PubMed]
- 108. Dreesen O. Towards delineating the chain of events that cause premature senescence in the accelerated aging syndrome Hutchinson-Gilford progeria (HGPS). Biochem Soc Trans. 2020; 48:981–91. https://doi.org/10.1042/BST20190882 [PubMed]
- 109. Filesi I, Gullotta F, Lattanzi G, D'Apice MR, Capanni C, Nardone AM, Columbaro M, Scarano G, Mattioli E, Sabatelli P, Maraldi NM, Biocca S, Novelli G. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005; 23:150–8. https://doi.org/10.1152/physiolgenomics.00060.2005 [PubMed]
- 110. Lattanzi G, Columbaro M, Mattioli E, Cenni V, Camozzi D, Wehnert M, Santi S, Riccio M, Del Coco R, Maraldi NM, Squarzoni S, Foisner R, Capanni C. Pre-Lamin A processing is linked to heterochromatin organization. J Cell Biochem. 2007; 102:1149–59. https://doi.org/10.1002/jcb.21467 [PubMed]
- 111. Whitton H, Singh LN, Patrick MA, Price AJ, Osorio FG, López-Otín C, Bochkis IM. Changes at the nuclear lamina alter binding of pioneer factor Foxa2 in aged liver. Aging Cell. 2018; 17:e12742. https://doi.org/10.1111/acel.12742 [PubMed]
- 112. Hildebrandt ER, Arachea BT, Wiener MC, Schmidt WK. Ste24p Mediates Proteolysis of Both Isoprenylated and Non-prenylated Oligopeptides. J Biol Chem. 2016; 291:14185–98. https://doi.org/10.1074/jbc.M116.718197 [PubMed]
- 113. Ast T, Michaelis S, Schuldiner M. The Protease Ste24 Clears Clogged Translocons. Cell. 2016; 164:103–14. https://doi.org/10.1016/j.cell.2015.11.053 [PubMed]
- 114. Kayatekin C, Amasino A, Gaglia G, Flannick J, Bonner JM, Fanning S, Narayan P, Barrasa MI, Pincus D, Landgraf D, Nelson J, Hesse WR, Costanzo M, et al, and AMP T2D-GENES Consortium. Translocon Declogger Ste24 Protects against IAPP Oligomer-Induced Proteotoxicity. Cell. 2018; 173:62–73.e9. https://doi.org/10.1016/j.cell.2018.02.026 [PubMed]
- 115. Spear ED, Hsu ET, Nie L, Carpenter EP, Hrycyna CA, Michaelis S. ZMPSTE24 missense mutations that cause progeroid diseases decrease prelamin A cleavage activity and/or protein stability. Dis Model Mech. 2018; 11:dmm033670. https://doi.org/10.1242/dmm.033670 [PubMed]
- 116. Fu B, Wang L, Li S, Dorf ME. ZMPSTE24 defends against influenza and other pathogenic viruses. J Exp Med. 2017; 214:919–29. https://doi.org/10.1084/jem.20161270 [PubMed]
- 117. Li S, Fu B, Wang L, Dorf ME. ZMPSTE24 Is Downstream Effector of Interferon-Induced Transmembrane Antiviral Activity. DNA Cell Biol. 2017; 36:513–7. https://doi.org/10.1089/dna.2017.3791 [PubMed]
- 118. Han M, Pandey D. ZMPSTE24 Regulates SARS-CoV-2 Spike Protein-enhanced Expression of Endothelial PAI-1. Am J Respir Cell Mol Biol. 2021; 65:300–8. https://doi.org/10.1165/rcmb.2020-0544OC [PubMed]
- 119. Khan SS. The Central Role of PAI-1 in COVID-19: Thrombosis and beyond. Am J Respir Cell Mol Biol. 2021; 65:238–40. https://doi.org/10.1165/rcmb.2021-0208ED [PubMed]
- 120. Davies BSJ, Barnes RH
2nd , Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG. An accumulation of nonfarnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genet. 2010; 19:2682–94. https://doi.org/10.1093/hmg/ddq158 [PubMed] - 121. Nacarelli T, Azar A, Sell C. Aberrant mTOR activation in senescence and aging: A mitochondrial stress response? Exp Gerontol. 2015; 68:66–70. https://doi.org/10.1016/j.exger.2014.11.004 [PubMed]
- 122. Nissan X, Blondel S, Navarro C, Maury Y, Denis C, Girard M, Martinat C, De Sandre-Giovannoli A, Levy N, Peschanski M. Unique preservation of neural cells in Hutchinson- Gilford progeria syndrome is due to the expression of the neural-specific miR-9 microRNA. Cell Rep. 2012; 2:1–9. https://doi.org/10.1016/j.celrep.2012.05.015 [PubMed]
- 123. Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BS, Yang SH, Barnes RH
2nd , Hong J, Sun T, Pleasure SJ, Young SG, Fong LG. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A. 2012; 109:E423–31. https://doi.org/10.1073/pnas.1111780109 [PubMed] - 124. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017; 16:203–22. https://doi.org/10.1038/nrd.2016.246 [PubMed]
- 125. Infante A, Gago A, de Eguino GR, Calvo-Fernández T, Gómez-Vallejo V, Llop J, Schlangen K, Fullaondo A, Aransay AM, Martín A, Rodríguez CI. Prelamin A accumulation and stress conditions induce impaired Oct-1 activity and autophagy in prematurely aged human mesenchymal stem cell. Aging (Albany NY). 2014; 6:264–80. https://doi.org/10.18632/aging.100651 [PubMed]
- 126. Ruiz de Eguino G, Infante A, Schlangen K, Aransay AM, Fullaondo A, Soriano M, García-Verdugo JM, Martín AG, Rodríguez CI. Sp1 transcription factor interaction with accumulated prelamin a impairs adipose lineage differentiation in human mesenchymal stem cells: essential role of sp1 in the integrity of lipid vesicles. Stem Cells Transl Med. 2012; 1:309–21. https://doi.org/10.5966/sctm.2011-0010 [PubMed]
- 127. Huang S, Risques RA, Martin GM, Rabinovitch PS, Oshima J. Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Exp Cell Res. 2008; 314:82–91. https://doi.org/10.1016/j.yexcr.2007.08.004 [PubMed]
- 128. di Masi A, D'Apice MR, Ricordy R, Tanzarella C, Novelli G. The R527H mutation in LMNA gene causes an increased sensitivity to ionizing radiation. Cell Cycle. 2008; 7:2030–7. https://doi.org/10.4161/cc.7.13.6149 [PubMed]
- 129. Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008; 10:452–9. https://doi.org/10.1038/ncb1708 [PubMed]
- 130. Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, Kennedy BK. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol Biol Cell. 2008; 19:5238–48. https://doi.org/10.1091/mbc.e08-05-0492 [PubMed]
- 131. Krishnan V, Chow MZ, Wang Z, Zhang L, Liu B, Liu X, Zhou Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci U S A. 2011; 108:12325–30. https://doi.org/10.1073/pnas.1102789108 [PubMed]