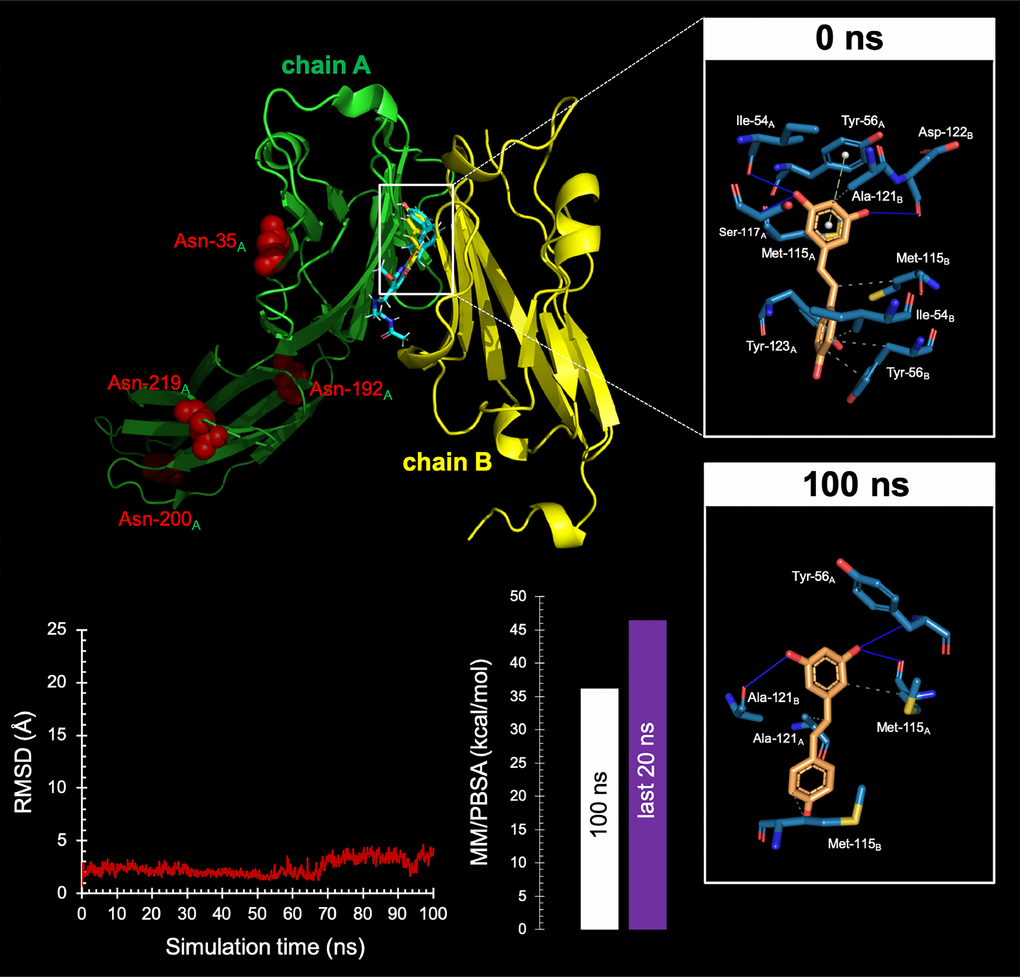
Figure 8.Resveratrol is predicted to bind the PD-1 dimer interface.Top Backbone representation of the PD-L1:PD-L1 dimer showing the computationally-predicted location of RSV (yellow) and BMS-202 (cyan). Chain A shows the location of the four Asn residues that can be glycosylated. The insets show the detailed maps of the molecular interactions of RSV with the amino acids at the hydrophobic pocket accommodating the BMS-202 inhibitor and formed at the PD-L1 dimer surface before (0 ns) and after 100 ns of molecular dynamics (MD) simulation, indicating the participating amino acids involved in the interaction and the type of interaction (hydrogen bonds, hydrophilic interactions, salt bridges, Π-stacking, etc). Bottom. Left. Trajectory of the RSV-forming complex with the PD-L1 dimer. Right. Molecular Mechanics/Poisson-Boltzmann Surface Area free energy analysis of the PD-L1 dimer forming a complex with RSV using YASARA dynamics v19.9.17 software. The best-docked complex as the initial conformation for MD simulation followed by 1000 snapshots (100 ns) obtained from the MD trajectory were employed to calculate the values of free energy binding of RSV. Additionally, the average value calculated for the last 200 snapshots (20 ns) is also displayed. YASARA-calculated binding energy provides positive values when the predicted binding is strong and stable whereas negative values indicate no binding. Figures were prepared using PyMol 2.3 software.