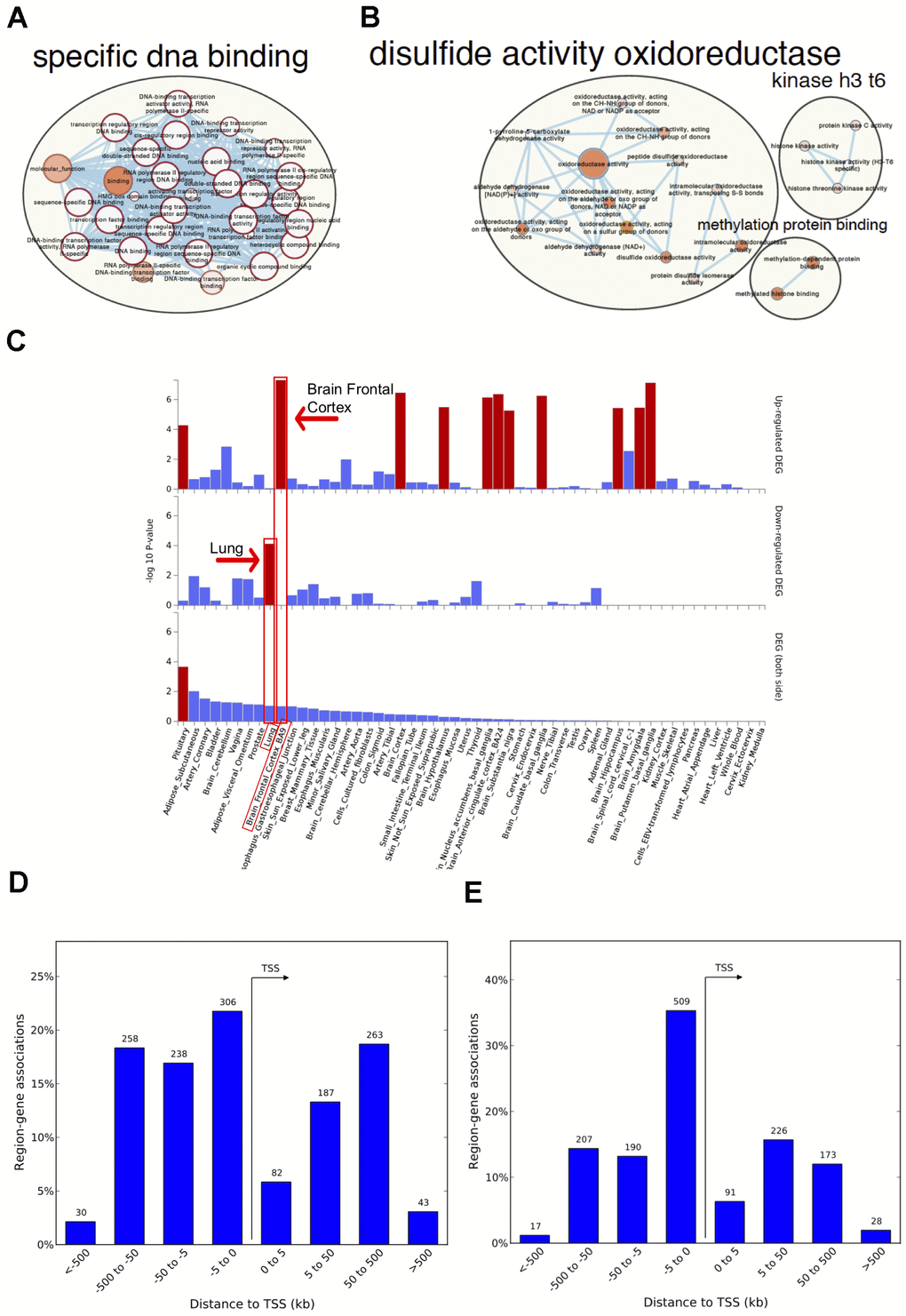
Figure 2.Network visualization, functional enrichment and region-gene associations in both fetal and adult lung tissue datasets. Network clusters of the molecular function gene ontology terms and annotated pathways including reactome and wikiPathways were created using the gene symbols mapped to the significant and age-associated differentially methylated positions (DMPs) overlapping between fetal and adult lung datasets. (A) Hyper-methylated CpGs were mainly enriched in transcription factor DNA binding whereas (B) hypo-methylated CpGs were enriched in oxidoreductase activity. Size corresponds to the overlap of genes between the enriched terms and color corresponds to significance. The analysis for hypo-methylated CpGs was limited by few numbers of genes represented by few CpGs (C) Functional enrichment of age-associated hyper-methylated DMPs in both fetal and adult lung tissue datasets amongst differentially expressed genes in 54 GTEx tissues (x-axis) and –log10(P-value) on the y-axis. Tissues with significant gene enrichment (FDR<0.05) are highlighted by red bars and tissues with the highest enrichment amongst the downregulated and upregulated genes are highlighted by arrows (D) Region-Gene associations using chromosomal coordinates for the differentially methylated regions in fetal lung dataset (E) Adult lung tissue dataset. TSS: transcription start site.