



The food we eat has long been linked to the rate we age. Selective pressures in times of food abundance and scarcity have influenced our very genetic makeup, instilling in our genome genes believed to control the delicate balance between metabolism and aging. However, this balance has been disrupted in western societies with developments in agriculture and technologies that have promoted the intake of high-calorie diets and sedentary lifestyles. We are witnessing an alarming increase in the rate of metabolic syndrome, which consists of a collection of abnormalities including obesity, type 2 diabetes, dyslipidemia, fatty liver, and a pro-inflammatory and prothrombotic state [1,2] Currently, one in four adults in the United States suffers from metabolic syndrome and worldwide estimates are over 2.1 billion [3,4]. Ultimately, this epidemic threatens human life-span projections and puts great pressure on our already overburdened health care system.
The sirtuin family of proteins appears to be at the crossroads between nutritional status and longevity. Sirtuins are highly conserved NAD+-dependent protein deacetylases and/or ADP ribosyltransferases that target histones, transcription factors, and co-regulators to adapt gene expression in response to the cellular energy state [5]. Many members of this family, including the founder Sir2, have been shown to impact aging in species ranging from yeast to fly and it is believed these protective actions result from the beneficial regulation of stress management, and energy homeostasis. SIRT1, the mammalian ortholog of Sir2, plays a role in numerous physiological processes including fat metabolism, glucose homeostasis and immune response. Because SIRT1 activity is dependent on the energy status of the cell, it provides a direct link between metabolism, chromosome structure, and metabolic gene regulation [6].
The liver is a central metabolic organ in charge of regulating nutrient homeostasis in fed and fasting conditions. It controls key aspects of lipid and glucose metabolism in response to nutritional and hormonal signals [7]. Tight regulation of glucose by the liver is essential to ensuring that glucose-dependent tissues such as brain and red blood cells have ample energy supply during periods of nutrient deprivation. Recent reports have shown that SIRT1 protein levels and enzymatic activity are induced in the fasted liver [8,9]. SIRT1 regulates genes involved in gluconeogenesis through deacetylation of several key transcription factors and coactivators [8,9,10]. The liver also plays an important role in maintaining lipid homeostasis. In line with its role as a metabolic mediator, SIRT1 is known to regulate genes involved in fatty acid oxidation and lipolysis [11]. Interestingly, the SIRT1 activator resveratrol has shown promise as a therapeutic agent for the treatment of metabolic diseases [12,13]. Mice fed a high-fat diet along with resveratrol remained lean and healthy compared to over-weight control animals [13]. Additionally, resveratrol significantly increased aerobic capacity, as evidenced by increased running time and elevated oxygen consumption in muscle fibers. Resveratrol treatment also protected mice against diet-induced-obesity and insulin resistance [12]. Groups are now focusing on the development of high affinity small molecule activators of SIRT1 as a therapeutic approach for treating diseases of aging such as type-2 diabetes [14].
Although SIRT1 is an important regulator of metabolism, the tissue-specific and systemic roles of SIRT1 are difficult to dissect in vivo, primarily due to the complicated developmental defects in the SIRT1 whole-body knockout mouse [15,16]. In search of further evidence to identify a tissue-specific role of SIRT1 in the regulation of energy homeostasis, we developed a knockout mouse model containing hepatic deletion of SIRT1 (LKO) [17]. Microarray analysis of liver from LKO mice revealed a striking reduction in expression of genes regulated by the peroxisome proliferators-activated receptors α (PPARα). This lipid sensing nuclear receptor is an important mediator of the adaptive response to fasting and starvation. Deletion of SIRT1 in the liver impairs PPARα signaling and decreases fatty acid β-oxidation, whereas over-expression of SIRT1 induces expression of PPARα target genes. Furthermore, we found that SIRT1 regulates PPARα signaling by directly interacting with the PPARα nuclear receptor. This interaction appears to be ligand dependent, as SIRT1 is recruited to response elements on promoters of PPARα target genes by agonists as well as by changes of nutritional status. One mechanism by which SIRT1 regulates PPARα signaling in the liver appears to be through the hands of PGC-1α, a key coactivator for PPARα signaling and a direct target of SIRT1 [9,18]. It has been shown that SIRT1 activates PGC-1α primarily by its deacetylation [9] (Figure 2). In keeping with these findings, we observed that although PGC-1α message levels are lower in SIRT1 LKO livers, PGC-1α protein accumulates on promoter regions of PPARα target genes but in a less active hyperacetylated form. These findings suggest that activated PGC-1α is required for promoting transcription of PPARα targets and that SIRT1 may be involved in monitoring the recruitment/dissociation cycle of PGC-1α. Additionally, GST-pull down mapping data showed that the core domain of SIRT1 directly interacts with PPARα. Therefore, another plausible mechanism underlying our observations is that PPARα may be a bona fide SIRT1 substrate. Further studies are necessary to elucidate weather SIRT1 indeed deacetylates PPARα, thereby affecting its activity.
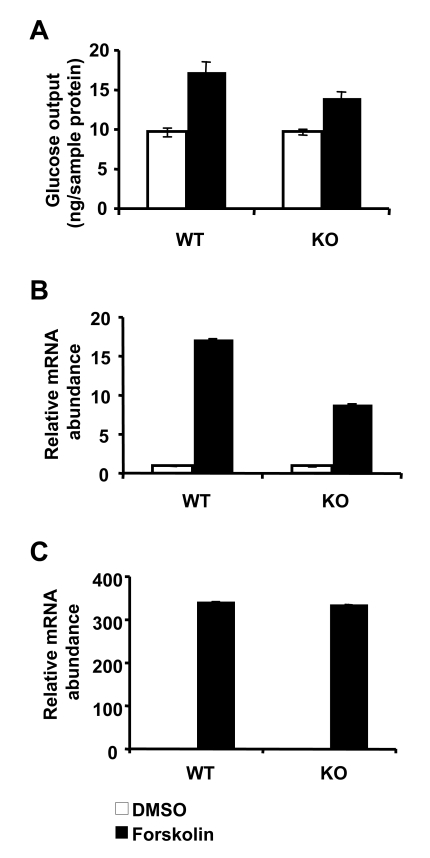
Figure 1. Loss of SIRT1 has minimal impact on gluconeogenesis in primary hepatocytes. (A) Glucose output
from primary hepatocytes isolated from control and SIRT1 LKO mice. Cells
were treated with DMSO (white bars) or 10 μM forskolin (black bars) and
incubated for 6 h in glucose free DMEM supplemented with 20 mM sodium
lactate and 2 mM sodium pyruvate. Glucose output was measured in culture
medium using a glucose oxidase kit (Sigma). Data represent mean +
SD. (B-C) SIRT1 deficiency in primary hepatocytes reduces the
induction of PGC-1α (B) but not
PEPCK (C) message in response to 10μM forskolin treatment. mRNA from
primary hepatocytes treated with DMSO (white bars) or forskolin (black
bars) were analyzed using qPCR. Data represent mean +
SD.
A major focus of our study was to characterize how disruptions in PPARα signaling affect the physiology of SIRT1 LKO mice [17]. When challenged with a high-fat diet, LKO mice displayed increased hepatic steatosis and hallmarks of endoplasmic reticulum stress and inflammatory responses. Interestingly, in a trend very similar to those reported in the PPARα knockout mouse, LKO mice displayed elevated levels of proinflammatory cytokines. These observations indicate that SIRT1 LKO mice are prone to development of hepatic inflammation, which has been implicated in the progression of insulin resistance [9,20]. These findings provide evidence that solidify SIRT1's role as a key regulator of metabolic homeostasis and complement previous animal studies using pharmacological tools [14] or modest SIRT1 overexpression mouse models [21,22].
Several of the metabolic abnormalities we observed in the SIRT1 LKO mice [17], however, are in direct contrast to those recently reported by Chen et al. [23].
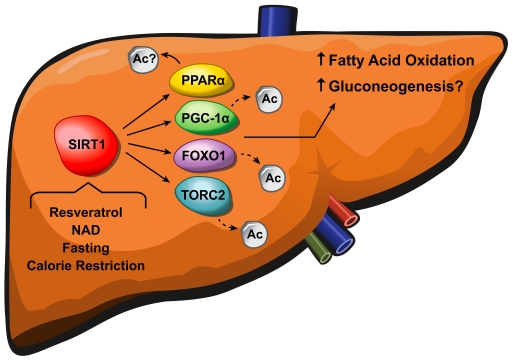
Figure 2. SIRT1 regulates fatty acid oxidation and gluconeogenesis in the liver. Resveratrol, NAD+,
fasting and calorie restriction activate SIRT1, causing deacetylation of
PGC-1α, FOXO1, and
TORC2 which in turn leads to increased fatty acid oxidation and
gluconeogenesis. The exact mechanism underlying how SIRT1 activates PPARα and the precise role of PGC-1α in the SIRT1-mediated glucose
homeostasis remain to be clarified.
Using a similar hepatic-specific knockout mouse model, Chen et al. observed a reduction in weight gain and liver fat accumulation in LKO mice when fed a western-style diet. Additionally, their mice were protected from the physiological impacts of a western diet with lower blood glucose and insulin levels. Similar to our study, their group observed minor physiological differences in LKO mice fed a chow diet. In wake of these findings, Chen et al. proposed that SIRT1 activity in the liver is directly proportional to calorie intake, and that excess calories and/or SIRT1 activators may result in elevated synthesis of fat and cholesterol. One possible factor contributing to the discrepancy between our observations and those of Chen et al. may be the difference in age of animals at which the feeding was initiated and data were collected. In our study, mice were six-week old when high-fat diet feeding was initiated, whereas four-month old mice were utilized in the study carried out by Chen et al. The varied responses of SIRT1 LKO mice to a western-style diet at different ages raises the possibility that hepatic SIRT1 may selectively regulate alternative metabolic pathways at multiple stages of development. An inducible SIRT1 knockout model will be helpful to dissect age-dependent effects of SIRT1. Moreover, since the liver is such a dynamic metabolic organ, small variations in dietetic components and genetic backgrounds may also contribute to the inconsistency between these two studies.
Another surprising phenotype observed in the SIRT1 LKO mice is their normal gluconeogenesis in response to a 16-h fasting [17]. The inducible coactivator PGC-1α is an important component of a number of transcriptional complexes that regulate glucose and lipid metabolism. Hepatic knockdown of SIRT1 significantly abrogates the fasting induction of gluconeogenic genes by regulating the acetylation status of PGC1α [11]. However, we observed no changes in fasting glucose levels in the absence of hepatic SIRT1 despite impaired PGC-1α signaling. Liver specific SIRT1 knockout mice had slightly higher, although not statistically significant, fasting glucose levels compared to littermate controls upon high-fat feeding. Expression levels of the two rate-limiting enzymes in the gluconeogenic pathway, PEPCK and G-6Pase, were also unchanged in the absence of hepatic SIRT1. Consistent with these observations, forskolin, an intracellular cAMP stimulator, promoted gluconeogenesis independently of SIRT1 levels in primary hepatocytes (Figure 1A). Additionally, although the forskolin-mediated induction of PGC1α expression was decreased in these cells (Figure 1B), the overall message levels of PEPCK remained similar between control and LKO hepatocytes (Figure 1C). Gluconeogenesis is regulated by a complex interplay between transcription factor and hormonal and coregulator signaling. While PGC-1α is known to control hepatic glucose production, other factors such as FOXO1 and TORC2 are reported to promote gluconeo-genesis [24]. Interestingly, SIRT1 has been shown to deacetylate and repress both FOXO1 [25] and TORC2 [24]. Therefore, a likely explanation for our findings is that while PGC-1α activity is lower in SIRT1 KO livers, compensatory effects of FOXO1 and TORC2 balance the reduction in PGC-1α signaling (Figure 2). Another possible explanation for the contradiction in these studies may lie in differences in cell types and method of SIRT1 deletion/knockdown used in the animal studies. It is important to note that the hepatic-specific albumin-Cre driven SIRT1 knockout mouse utilized in our study is a permanent knockout model. Phenotypes observed in these mice may reflect systemic and local compensatory effects in wake of hepatic deletion of SIRT1. Studies done by Rodger et al. [11] employed transient knockdown methods using adenovirus-mediated shRNA which seem to provoke more acute responses to loss of hepatic SIRT1.
In conclusion, while our study defines a new role for SIRT1 as a key regulator of hepatic lipid metabolism, it also adds fuel to the fire of controversy surrounding this protein as a central player in mammalian energy homeostasis. It appears that in the liver, a major target of this sirtuin is the PPARα/PGC-1α signaling axis. Ablation of SIRT1 in the liver creates disruptions in fatty acid oxidation, increased cellular stress, and elevations in proinflammatory cytokines. What remains to be determined is the precise role SIRT1 plays in regulating gluconeogenesis and cholesterol metabolism in the liver and how this, in turn, affects systemic metabolism. Our findings and others suggest that activation of SIRT1 may provide a therapeutic strategy for treatment of metabolic syndrome.
Acknowledgments
We thank Drs. Sailesh Surapureddi and Anton Jetten for criticalreading of the manuscript; Dr. Frederic Alt at Harvard Medical School for providing the SIRT1 exon 4 floxed allele; and NIEHS Multimedia Services Department for the cartoon graph of Figure 2. This work was supported by the IntramuralResearch Program of the NIH, National Institute of EnvironmentalHealth Sciences to X.L. (Z01 ES102205).
Conflicts of Interest
The authors in this manuscript have no conflict of interests to declare.
References
- 1. Eckel RH , Grundy SM and Zimmet PZ. The metabolic syndrome. Lancet. 2005; 65: 1415 -28. [PubMed] .
- 2. Grundy SM , HB Brewer Jr , Cleeman JI , Smith SC Jr and Lenfant C. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Arterioscler Thromb Vasc Biol. 2004; 24: e13 -e18. [PubMed] .
- 3. Flegal KM , Carroll MD , Ogden CL and Johnson CL. Prevalence and trends in obesity among US adults, 1999-2000. Jama. 2002; 288: 1723 -1727. [PubMed] .
- 4. Li Z , Bowerman S and Heber D. Health ramifications of the obesity epidemic. Surg Clin North Am. 2005; 85: 681 -701. [PubMed] .
- 5. Blander G and Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004; 73: 417 -435. [PubMed] .
- 6. Bishop NA and Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007; 8: 835 -844. [PubMed] .
- 7. van den Berghe G The role of the liver in metabolic homeostasis: implications for inborn errors of metabolism. J Inherit Metab Dis. 1991; 14: 407 -420. [PubMed] .
- 8. Liu Y , Dentin R , Chen D , Hedrick S , Ravnskjaer K , Schenk S , Milne J , Meyers JD , Cole P , Iii JY , Olefsky J , Guarente L and Montminy M. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008; 456: 269 -273. [PubMed] .
- 9. Rodgers JT , Lerin C , Haas W , Gygi SP , Spiegelman BM and Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005; 434: 113 -118. [PubMed] .
- 10. Motta MC , Divecha N , Lemieux M , Kamel C , Chen D , Gu W , Bultsma Y , McBurney M and Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004; 116: 551 -563. [PubMed] .
- 11. Rodgers JT and Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A. 2007; 104: 12861 -12866. [PubMed] .
- 12. Baur JA , Pearson KJ , Price NL , Jamieson HA , Lerin C , Kalra A , Prabhu VV , Allard JS , Lopez-Lluch G , Lewis K , Pistell PJ and Poosala S. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006; 444: 337 -342. [PubMed] .
- 13. Lagouge M , Argmann C , Gerhart-Hines Z , Meziane H , Lerin C , Daussin F , Messadeq N , Milne J , Lambert P , Elliott P , Geny B , Laakso M , Puigserver P and Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006; 127: 1109 -1122. [PubMed] .
- 14. Milne JC , Lambert PD , Schenk S , Carney DP , Smith JJ , Gagne DJ , Jin L , Boss O , Perni RB , Vu CB , Bemis JE and Xie R. et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007; 450: 712 -716. [PubMed] .
- 15. Cheng HL , Mostoslavsky R , Saito S , Manis JP , Gu Y , Patel P , Bronson R , Appella E , Alt FW and Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003; 100: 10794 -10799. [PubMed] .
- 16. McBurney MW , Yang X , Jardine K , Hixon M , Boekelheide K , Webb JR , Lansdorp PM and Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003; 23: 38 -54. [PubMed] .
- 17. Purushotham A , Schug TT , Xu Q , Surapureddi S , Guo X and Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009; 9: 327 -338. [PubMed] .
- 18. Li S , Liu C , Li N , Hao T , Han T , Hill DE , Vidal M and Lin JD. Genome-wide coactivation analysis of PGC-1alpha identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab. 2008; 8: 105 -117. [PubMed] .
- 19. Kanda H , Tateya S , Tamori Y , Kotani K , Hiasa K , Kitazawa R , Kitazawa S , Miyachi H , Maeda S , Egashira K and Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006; 116: 1494 -1505. [PubMed] .
- 20. Weisberg SP , McCann D , Desai M , Rosenbaum M , Leibel RL and Ferrante Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003; 112: 1796 -1808. [PubMed] .
- 21. Banks AS , Kon N , Knight C , Matsumoto M , Gutierrez-Juarez R , Rossetti L , Gu W and Accili D. SirT1 Gain of Function Increases Energy Efficiency and Prevents Diabetes in Mice. Cell metabolism. 2008; 8: 333 -341. [PubMed] .
- 22. Pfluger PT , Herranz D , Velasco-Miguel S , Serrano M and Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008; 105: 9793 -9798. [PubMed] .
- 23. Chen D , Bruno J , Easlon E , Lin SJ , Cheng HL , Alt FW and Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008; 22: 1753 -1757. [PubMed] .
- 24. Liu Y , Dentin R , Chen D , Hedrick S , Ravnskjaer K , Schenk S , Milne J , Meyers DJ , Cole P , Iii JW , Olefsky J , Guarente L and Montminy M. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008; 456: 269 -273. [PubMed] .
- 25. Motta MC , Divecha N , Lemieux M , Kamel C , Chen D , Gu W , Bultsma Y , McBurney M and Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004; 116: 551 -563. [PubMed] .