What determines the switch between atrophic and neovascular forms ofage related macular degeneration? - the role of BMP4 induced senescence
Abstract
Age-related macular degeneration (AMD), the leadingcause of blindness in the elderly, targets the retinal pigment epithelium(RPE), a monolayer of cells at the back of the eye. As AMD progresses, itcan develop into two distinct forms of late AMD: "dry," atrophic AMD,characterized by RPE senescence and geographic RPE loss, and "wet,"neovascular AMD, characterized by RPE activation with abnormal growth ofchoroidal vessels. The genetic and molecular pathways that lead to thesediverse phenotypes are currently under investigation. We have found thatbone morphogenetic protein-4 (BMP4) is differentially expressed in atrophicand neovascular AMD. In atrophic AMD, BMP4 is highly expressed in RPE, andmediates oxidative stress induced RPE senescencein vitro via Smadand p38 pathways. In contrast, in neovascular AMD lesions, BMP4 expressionin RPE is low, possibly a result of local expression of pro-inflammatorymediators. Thus, BMP4 may be involved in the molecular switch determiningwhich phenotypic pathway is taken in the progression of AMD.
Age-related macular
degeneration (AMD) is the leading cause of irreversible blindness in the
elderly [1-2]. Considerable evidence supports the opinion that the retinal
pigment epithelium (RPE), a monolayer of cells between the light sensitive
photoreceptors and the vascular choroid, is a primary site of pathology in the
disease [1-5]. The RPE provides support for the photoreceptors and plays a
critical role in the visual cycle; thus, degeneration and loss of RPE lead to
secondary degeneration of photoreceptor cells [3]. Early AMD is characterized
by the presence of extracellular deposits, or drusen, beneath the RPE.
Increasing numbers of large drusen predispose to the development of the late blinding forms of the disease which can
manifest in two disparate ways. In late, "dry" AMD,
geographic loss of RPE occurs in the macular region, while in the late
neovascular or "wet" form of the disease, there is abnormal growth of choroidal
vessels under the retina which leak fluid and may progress to form a disciform
scar (Figure 1) [1-5]. Pathogenic mechanisms for AMD include both genetic and
environmentalfactors related to primary RPE senescence, alterations
in the complement pathway, increased inflammation, changes in the balance of
growth factors, excessive lipofuscinaccumulation, and oxidative
stress [5]. Major genetic risk factors for AMD, including Complement Factor H
and HTRA1 variants, appear to predispose to both atrophic and neovascular AMD
[6,7]; only recently has a genetic variant been identified that specifically
predisposes to the atrophic form [8]. Consequently, there is considerable
interest in further establishing the factors that mediate the "molecular
switch" that may determine which late form of the disease an individual
develops.
Recently, we reported that bone
morphogenetic protein (BMP)4 is prominently expressed in the RPE and adjacent
extracellular matrix of patients with the dry or atrophic form of AMD when
compared to controls (Figure 2A, B). Here, we show that in the wet or
neovascular form of the disease (5 patients with surgical excision of choroidal
neovascular membranes due to neovascular AMD) there is almost no expression of
BMP4 in the RPE and adjacent neovascular tissues (Figure 2C). Interestingly, in
cases (3 patients) in which the neovascular lesion had progressed to a fibrous
scar, the level of BMP4 expression increased in the RPE and adjacent tissues
(Figure 2D). This has led us to the hypothesis that BMP4 may be a molecular
switch participating in the pathway decision that
determines which form of late AMD develops.
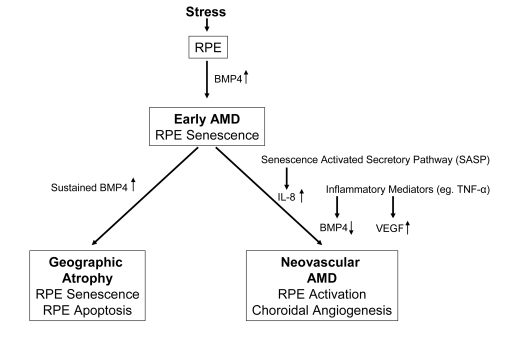
Figure 1. Diagram
illustrating the progression of early age related macular degeneration
(AMD) into 2 divergent late stages and the potential role of BMP4 as a
switch between these pathways. Chronic stressors such as oxidative stress
can promote the expression of BMP4 in the retinal pigment epithelium (RPE)
and induce RPE senescence as part of the phenotype of early AMD. If BMP4
expression is sustained, it could lead to RPE apoptosis and geographic
atrophy. In other individuals, activation of the senescence activated
secretory pathway and expression of pro-inflammatory mediators could result
in increased expression of interleukin (IL)-8, decreased expression of BMP4
and increased expression of vascular endothelial growth factor (VEGF)
resulting in neovascular AMD with choroidal angiogenesis.
BMP4 is an important regulator of
differentiation, senescence and apoptosis in many different cells and tissues
[9,10]. We reported that BMP4 can induce RPE senescence in vitro [11],
and that RPE chronically exposed to sublethal doses of oxidative stress can
increase their BMP4 expression and exhibit a senescent phenotype, thus
supporting the contention that BMP4 mediates oxidative stress-induced RPE
senescence. We further determined that BMP4 mediates RPE senescence via
activation of Smad and p38 pathways to activate p53, and increase expression of
p21WAF1/ cip1, and to decrease phospho-Rb. Importantly,
BMP4-mediated RPE senescence can be inhibited by Chordin-like, a BMP4
antagonist, and SB203580, a phospho-p38 inhibitor. Our findings not only
disclose a molecular pathway linking oxidative stress with RPE senescence, but
also provide a novel therapeutic target for treatment of atrophic AMD.
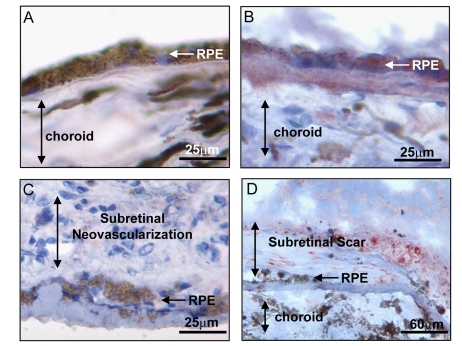
Figure 2.
Expression of BMP4 in late stages of age related macular degeneration
(AMD). Immunohistochemical stains for BMP4 (red chromogen) in retinal
pigment epithelium (RPE)/choroid tissue sections from donor eyes with
hematoxylin counterstain. In (A) a control individual without AMD
shows no apparent BMP4 staining in RPE or choroid. In (B) an
individual with late dry AMD, away from a region of geographic atrophy
shows prominent BMP4 immunoreactivity in RPE and in the accumulated drusen
material between the RPE and the choroid. In (C) an individual with
neovascular form of late AMD shows no apparent BMP4 staining in the RPE or
the neovascular lesion between the RPE and retina. In (D) an
individual with neovascular form of late AMD that further progressed to
scar with loss of neovascular channels shows re-expression of BMP4 staining
in cells within and adjacent to the lesion. Note loss of most cells in RPE
layer. The institutional review board (IRB) of
the University of Southern California approved our use of human donor eyes.
All procedures conformed to the Declaration of Helsinki forresearch
involving human subjects.
Recently, Demidenko et al. [12] evaluated
the concept that duration of cell cycle arrest determines the progressive loss
of proliferative capacity characteristic of cellular senescence [12]. Using a
variety of cell lines including the spontaneously immortalized human RPE cell
line, ARPE-19, they found that rapamycin, an inhibitor of the nutrient-sensor
mammalian target of rapamycin (mTOR), partially prevented loss of proliferative
potential induced by oxidative stress, or ectopic p21 or p16 exposure, leading
to deceleration of cellular senescence [12]. This work supports the
critical role of oxidative stress, and cell cycle arrest in induction of
senescence and demonstrates a pharmacologic approach to suppression of RPE
senescence [12].
Interestingly, BMP4 has been
found to be involved in chemotherapeutic
agent-induced premature senescence of cancer cells [13]. Adriamycin and BMP4
treatment can induce lung cancer cell senescence, and BMP4 expression is increased in Adriamycin-treated lung
cancer cells. This BMP4-induced premature senescence is mediated through Smad
signaling to up-regulate p16INK4a and p21WAF1/ cip1.
BMP4 and other BMP signaling pathways were also found to participate in
senescence of multiple cancer cell types or in the inhibition of tumor cell
growth [14,15]. For example, BMP-2 and -4 inhibit prostate cancer cell growth
through Smad-1 phosphorylation, p21WAF1/ cip1 up-regulation, and Rb
dephosphorylation, while in glioblastoma, BMP4 and its cognate receptors can
trigger the Smad signaling cascade to reduce the proliferation of tumor cells
[16]. Together, these studies reveal that BMP4 induces and mediates the
premature senescence of both malignant cells in tumors and aging RPE cells in
dry AMD.
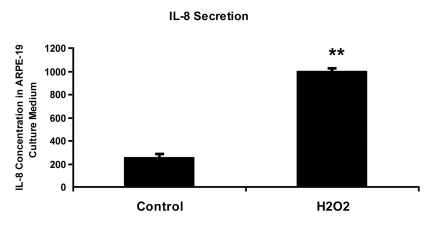
Figure 3.
IL-8 protein concentration in culture medium measured by ELISA. ARPE-19
cells were treated with 150 uM H2O2 in culture medium
with 10% fetal bovine serum for 2 hours and allowed to recover in stressor-free
ARPE medium for 22 hours. The procedure was repeated to generate the next
treatment cycle. The
twice treated cells were allowed to stay in 1% serum ARPE medium for 72
hours after stress before proceeding to further analytic assays.
The culture media from control and senescent RPE cells were collected and
used directly for ELISA measurement. IL-8 secretion level was measured in pg/ml using
human IL-8 ELISA Kit (BioLegend, Inc., San Diego, CA) according to
manufacturer's instructions. The level of IL-8 secretion shown here was
averaged from a triplicate of each sample and from 3 independent repeats of
H2O2 treatments. Student's t test was used for
statistical analysis (**; p < 0.0005).
Transforming growth factor (TGF)-β has been extensively reported to be
involved in mediating oxidative stress induced premature senescence of
fibroblasts [17-19]. Recently it has been reported that TGF-β mediates oxidative stress induced RPE
cell senescence through the up-regulation of p21 WAF1/ cip1 and the
down-regulation of phosphorylated Rb and that blockade of TGF-β signaling by specific TGF-β antibody can impede RPE senescence [20].
This finding is similar to our finding for BMP4 mediated oxidative
stress-induced RPE senescence. We suggest that TGF-β and BMP4 may have a synergistic effect in
mediating the oxidative stress-induced RPE senescence, because neither TGF-β antibodies nor BMP4 antagonist alone can
completely block the expression of senescence marker genes to baseline in the oxidative
stress treated RPE cells. More investigations are needed to elucidate the
interactions between TGF-β and BMP
signaling cascades in oxidative stress-induced RPE senescence.
A variety of intrinsic and
extrinsic stress signals can activate the p53 pathway, which then triggers
either cellular senescence or apoptosis [21,22]. We found that both BMP4 and oxidant
treatment can increase p53 protein level in RPE cells. A microarray analysis of
the RPE transcriptome from the maculas of six healthy, elderly human donors
revealed a statistically significant overrepresentation of genes associated
with stress, with the p53 gene listed in the
top 30 most highly expressed RPE genes [23].
Although little is known about how p53 regulates cellular senescence and how
p53 interacts with the BMP-Smad pathway, the fact that p53 levels were
increased in RPE cells after BMP4 treatment and Smad1/5 could bind to p53 [24],
raises the possibility that Smad1/5 activates p53 dependent transcription through
the regulation of post-translational modifications of p53, such as
phosphorylation and acetylation.
It remains
unanswered why some patients develop atrophic AMD while others develop the
neovascular form of the disease. The switch between dry and wet AMD may be
related to differences in the microenvironment created by senescent RPE cells,
which secrete a number of cytokines and growth factors [25]. The defined
components of this "senescence associated secretory phenotype" (SASP) include
elements associated with inflammation, and angiogenesis, such as interleukin
(IL)-6 and IL-8 [26-28]. We have found that RPE cells induced into senescence
by chronic oxidative stress secrete 4 times higher IL-8 than non-senescent RPE
cells (Figure 3). IL-8 promotes angiogenesis by increasing the proliferation, survival and
migration of endothelial cells and promotes inflammation by increasing
neutrophil chemotaxis and degranulation [29-31]. Together these findings
suggest that chronic oxidative stress increases the premature senescence of
RPE. If RPE do not go down the cell death
pathway to atrophic AMD, the senescent RPE may secrete high levels of IL-8,
which in turn stimulate inflammation and angiogenesis. But what about the
finding that neovascular AMD lesions show minimal levels of BMP4? In other cell
types, pro-inflammatory mediators such as tumor necrosis factor (TNF)-alpha
have been shown to downregulate BMP4 expression [32]. In the absence of BMP4,
neovascular endothelial cells, stimulated by increased expression of vascular
endothelial growth factor, and without the growth inhibitory senescence and
cell death effects mediated by BMP4, would be in a permissive environ-ment for
angiogenesis [5]. This idea is further supported by the finding that when
neovascular AMD lesions undergo subsequent scar formation, with degeneration
and loss of neovascular endothelial cells, there is a concomitant increase in
BMP4 expression (Figure 2).
It has been previously observed that
tissues in aged individuals may exhibit the paradoxical juxtaposition of
atrophy and hyperplasia within the same tissue or even within the same cell
type [33]. This response may be explained in part by senescent heterogeneity
[34,35]. In vitro culture of human fibroblasts results in a fraction of
cells senescing at every population doubling. The senescent cells have shorter
telomeres than their cycling counterparts. Thus, it was concluded that the main
cause of intrinsic heterogeneity of senescent fibroblasts was the cell to cell
variation of telomere shortening [36]. Using pulse-chase
5-bromodeoxyuridine-labeling assay, Gonzalez and colleagues revealed that the
senescent heart contained functionally competent cardiac progenitor cells
(CPCs) with longer telomeres, and these stem cell-like CPCs can be activated
and migrate to the damaged regions to generate a population of young
cardiomyocytes and partly reverse the aging myopathy [37].
Much remains to be learned about the
genetic and environmental factors mediating the progression of early AMD to its
late forms. Our finding of differential expression of BMP4 in geographic
atrophy and neovascular AMD and the interactive roles of oxidative stress,
inflammation and senescence in the regulation and functional effects of this
growth factor, suggests the possibility that BMP4 may be playing a part in the
molecular switch determining which phenotypic pathway is taken in the
progression of AMD.
Acknowledgments
This work was supported by National Institutes of
Health grants EY01545 and EY03040 and by the Arnold and Mabel Beckman
Foundation.
Conflicts of Interest
The authors of this manuscript have no
conflict of interest to declare.
References
-
1.
Zarbin
MA
Current concepts in the pathogenesis of age-related macular degeneration.
Arch Ophthalmol.
2004;
122:
598
-614.
[PubMed]
.
-
2.
Ambati
J
, Ambati
BK
, Yoo
SH
, Ianchulev
S
and Adamis
AP.
Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies.
Surv Ophthalmol.
2003;
48:
257
-293.
[PubMed]
.
-
3.
Holz
FG
, Pauleikhoff
D
, Klein
R
and Bird
AC.
Pathogenesis of lesions in late age-related macular disease.
American journal of ophthalmology.
2004;
137:
504
-510.
[PubMed]
.
-
4.
Klein
ML
, Ferris
FL 3rd
, Armstrong
J
, Hwang
TS
, Chew
EY
, Bressler
SB
and Chandra
SR.
Retinal precursors and the development of geographic atrophy in age-related macular degeneration.
Ophthalmology.
2008;
115:
1026
-1031.
[PubMed]
.
-
5.
Ding
X
, Patel
M
and Chan
CC.
Molecular pathology of age related macular degeneration.
Prog Retin Eye Res.
2009;
28:
1
-18.
[PubMed]
.
-
6.
Cameron
DJ
, Yang
Z
, Gibbs
D
, Chen
H
, Kaminoh
Y
, Jorgensen
A
, Zeng
J
, Luo
L
, Brinton
G
, Brand
JM
, Bernstein
PS
, Zabriskie
NA
, Tang
S
, Constantine
R
, Tong
Z
and Zhang
K.
HTRA1 variant confers similar risks to geographic atrophy and neovascular age-related macular degeneration.
Cell Cycle.
2007;
6:
1122
-1125.
[PubMed]
.
-
7.
Sepp
T
, Khan
JC
, Thurlby
DA
, Shahid
H
, Clayton
DG
, Moore
AT
, Bird
AC
and Yates
JR.
Complement factor H variant Y402H is a major risk determinant for geographic atrophy and choroidal neovascularization in smokers and nonsmokers.
Invest Ophthalmol Vis Sci.
2006;
47:
536
-540.
[PubMed]
.
-
8.
Yang
Z
, Stratton
C
, Francis
P
, Kleinman
ME
, Tan
PL
, Gibbs
D
, Tong
Z
, Chen
H
, Constantine
R
, Yang
X
, Chen
Y
, Zeng
J
, Davey
L
, Ma
X
, Hau
VS
, Wang
C
, Harmon
J
, Buehler
J
, Pearson
E
, Patel
S
, Kaminoh
Y
, Watkins
S
, Luo
L
, Zabriskie
NA
, Berstein
PS
, Cho
W
, Schwager
A
, Hinton
DR
, Klein
ML
, Hamon
SC
, Simmons
E
, Sunness
JS
, Campochiaro
P
, Jorde
L
, Parmigiani
G
, Zack
DL
, Katsanis
N
, Ambati
J
and Zhang
K.
Toll-Like Receptor-3 and Geographic Atrophy in Age-Related Macular Degeneration.
New Engl J Med.
2008;
359:
1456
-63.
[PubMed]
.
-
9.
Yokouchi
Y
, Sakiyama
J
, Kameda
T
, Iba
H
, Suzuki
A
, Ueno
N
and Kuroiwa
A.
BMP-2/-4 mediate programmed cell death in chicken limb buds.
Development.
1996;
122:
3725
-3734.
[PubMed]
.
-
10.
Trousse
F
, Esteve
P
and Bovolenta
P.
Bmp4 mediates apoptotic cell death in the developing chick eye.
J Neurosci.
2001;
21:
1292
-1301.
[PubMed]
.
-
11.
Zhu
D
, Wu
J
, Spee
C
, Ryan
SJ
and Hinton
DR.
BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration.
J Biol Chem.
2009;
284:
9529
-9539.
[PubMed]
.
-
12.
Demidenko
ZN
, Zubova
SG
, Bukreeva
EI
, Pospelov
VA
, Pospelova
TV
and Blagosklonny
MV.
Rapamycin decelerates cellular senescence.
Cell Cycle.
2009;
8:
1888
-1895.
[PubMed]
.
-
13.
Su
D
, Zhu
S
, Han
X
, Feng
Y
, Huang
H
, Ren
G
, Pan
L
, Zhang
Y
, Lu
J
and Huang
B.
BMP4-Smad signaling pathway mediates adriamycin-induced premature senescence in lung cancer cells.
J Biol Chem.
2009;
284:
12153
-12164.
[PubMed]
.
-
14.
Brubaker
KD
, Corey
E
, Brown
LG
and Vessella
RL.
Bone morphogenetic protein signaling in prostate cancer cell lines.
J Cell Biochem.
2004;
91:
151
-160.
[PubMed]
.
-
15.
Fukuda
N
, Saitoh
M
, Kobayashi
N
and Miyazono
K.
Execution of BMP-4-induced apoptosis by p53-dependent ER dysfunction in myeloma and B-cell hybridoma cells.
Oncogene.
2006;
25:
3509
-3517.
[PubMed]
.
-
16.
Piccirillo
SG
, Reynolds
BA
, Zanetti
N
, Lamorte
G
, Binda
E
, Broggi
G
, Brem
H
and Olivi
A.
Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells.
Nature.
2006;
444:
761
-765.
[PubMed]
.
-
17.
Frippiat
C
, Chen
QM
, Zdanov
S
, Magalhaes
JP
, Remacle
J
and Toussaint
O.
Subcytotoxic H2O2 stress triggers a release of transforming growth factor-beta 1, which induces biomarkers of cellular senescence of human diploid fibroblasts.
J Biol Chem.
2001;
276:
2531
-2537.
[PubMed]
.
-
18.
Chretien
A
, Dierick
JF
, Delaive
E
, Larsen
MR
, Dieu
M
, Raes
M
, Deroanne
CF
and Roepstorff
P.
Role of TGF-beta1-independent changes in protein neosynthesis, p38alphaMAPK, and cdc42 in hydrogen peroxide-induced senescence-like morphogenesis.
Free Radic Biol Med.
2008;
44:
1732
-1751.
[PubMed]
.
-
19.
Zdanov
S
, Remacle
J
and Toussaint
O.
Establishment of H2O2-induced premature senescence in human fibroblasts concomitant with increased cellular production of H2O2.
Ann N Y Acad Sci.
2006;
1067:
210
-216.
[PubMed]
.
-
20.
Yu
AL
, Fuchshofer
R
, Kook
D
, Kampik
A
, Bloemendal
H
and Welge-Lussen
U.
Subtoxic oxidative stress induces senescence in retinal pigment epithelial cells via TGF-beta release.
Invest Ophthalmol Vis Sci.
2009;
50:
926
-935.
[PubMed]
.
-
21.
Vousden
KH
and Lane
DP.
p53 in health and disease.
Nat Rev Mol Cell Biol.
2007;
8:
275
-283.
[PubMed]
.
-
22.
Harris
SL
and Levine
AJ.
The p53 pathway: positive and negative feedback loops.
Oncogene.
2005;
24:
2899
-2908.
[PubMed]
.
-
23.
Booij
JC
, van
Soest S
, Swagemakers
SM
, Essing
AH
, Verkerk
AJ
and van der Spek
PJ.
Functional annotation of the human retinal pigment epithelium transcriptome.
BMC Genomics.
2009;
10:
164
[PubMed]
.
-
24.
Takebayashi-Suzuki
K
, Funami
J
, Tokumori
D
, Saito
A
, Watabe
T
, Miyazono
K
, Kanda
A
and Suzuki
A.
Interplay between the tumor suppressor p53 and TGF beta signaling shapes embryonic body axes in Xenopus.
Development.
2003;
130:
3929
-3939.
[PubMed]
.
-
25.
Zender
L
and Rudolph
KL.
Keeping your senescence cells under control.
Aging.
2009;
1:
1
-4.
[PubMed]
.
-
26.
Koch
AE
, Polverini
PJ
, Kunkel
SL
, Harlow
LA
, DiPietro
LA
, Elner
SG
and Strieter
RM.
Interleukin-8 as a macrophage-derived mediator of angiogenesis.
Science.
1992;
258:
1798
-1801.
[PubMed]
.
-
27.
Yoshimura
T
, Matsushima
K
, Tanaka
S
, Robinson
EA
, Appella
E
, Oppenheim
JJ
and Leonard
EJ.
Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines.
Proc Natl Acad Sci U S A.
1987;
84:
9233
-9237.
[PubMed]
.
-
28.
Kuilman
T
, Michaloglou
C
, Vredeveld
LC
, Douma
S
, van
Doorn R
, Desmet
CJ
, Aarden
LA
, Mooi
WJ
and Peeper
DS.
Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network.
Cell.
2008;
133:
1019
-1031.
[PubMed]
.
-
29.
Coppe
JP
, Patil
CK
, Rodier
F
, Sun
Y
, Munoz
DP
, Goldstein
J
, Nelson
PS
, Desprez
PY
and Campisi
J.
Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor.
PLoS biology.
2008;
6:
2853
-2868.
[PubMed]
.
-
30.
Malgorzata
Goczalik I
, Raap
M
, Weick
M
, Milenkovic
I
, Heidmann
J
, Enzmann
V
, Wiedemann
P
, Reichenbach
A
and Francke
M.
The activation of IL-8 receptors in cultured guinea pig Muller glial cells is modified by signals from retinal pigment epithelium.
Journal of neuroimmunology.
2005;
161:
49
-60.
[PubMed]
.
-
31.
Waugh
DJ
and Wilson
C.
The interleukin-8 pathway in cancer.
Clin Cancer Res.
2008;
14:
6735
-6741.
[PubMed]
.
-
32.
Zhu
NL
, Li
C
, Huang
HH
, Sebald
M
, Londhe
VA
, Heisterkamp
N
, Warburton
D
, Bellusci
S
and Minoo
P.
TNF-alpha represses transcription of human bone morphogenetic protein-4 in lung epithelial cells.
Gene.
2007;
393:
70
-80.
[PubMed]
.
-
33.
Martin
GM
The genetics and epigenetics of altered proliferative homeostasis in ageing and cancer.
Mech Ageing Dev.
2007;
128:
9
-12.
[PubMed]
.
-
34.
Kill
IR
, Faragher
RG
, Lawrence
K
and Shall
S.
The expression of proliferation-dependent antigens during the lifespan of normal and progeroid human fibroblasts in culture.
Journal of cell science.
1994;
107(Pt 2):
571
-579.
[PubMed]
.
-
35.
Kirkwood
TB
and Finch
CE.
Ageing: the old worm turns more slowly.
Nature.
2002;
419:
794
-795.
[PubMed]
.
-
36.
Martin-Ruiz
C
, Saretzki
G
and Petrie
J.
Stochastic variation in telomere shortening rate causes heterogeneity of human fibroblast replicative life span.
J Biol Chem.
2004;
279:
17826
-17833.
[PubMed]
.
-
37.
Gonzalez
A
, Rota
M
, Nurzynska
D
, Misao
Y
, Tillmanns
J
, Ojaimi
C
, Padin-Iruegas
ME
, Muller
P
, Esposito
G
, Bearzi
C
, Vitale
S
, Dawn
B
, Sanganalmath
SK
, Baker
M
, Hintze
TH
, Bolli
R
, Urbanek
K
, Hosoda
T
, Anversa
P
, Kajstura
J
and Leri
A.
Activation of cardiac progenitor cells reverses the failing heart senescent phenotype and prolongs lifespan.
Circ Res.
2008;
102:
597
-606.
[PubMed]
.