



Aging involves multiple processes that render cells, tissues and organs vulnerable to stress, damage and ultimately death. Aging itself is not a disease, but there are a number of diseases that become exponentially more prevalent with advancing age such as cancer, cardiovascular disease, metabolic syndrome and neurodegenerative diseases. Energy sensing, food intake and caloric utilization must be kept in equilibrium to preserve appropriate fat stores to prevent the deregulation of glucose homeostasis and other obesity-related disorders.
Calorie restriction (CR) is an intervention aimed to produce undernutrition without malnutrition. CR increases healthspan and lifespan in almost all species tested such as yeast, insects, nematodes and mammals [1], including nonhuman primates [2]. CR has been studied extensively with consistent results showing its beneficial effects on longevity, age-associated diseases, attenuation of functional decline, and carcinogenesis across a variety of species and diet formulations [3]. Among mammals mice have been the most heavily-researched model with CR eliciting myriad behavioral, physiological, and metabolic changes that include decreased body temperature, blood glucose, insulin and fat mass, and increased physical activity, glucose tolerance and insulin sensitivity [4]. Studies in Saccharomyces cerevisiae and Drosophila melanogaster have demonstrated that SIR2, which encodes for a NAD+-dependent histone deacetylase, plays a central role in mediating the increase in longevity associated with CR in these species [5-7]. The involvement of SIR2 in lifespan extension by CR may relate to its responsiveness to nicotinamide levels and the NAD+/NADH ratio, both indicators of cellular energy status [8-10]. A growing body of evidence indicates that the mammalian homologue of SIR2, SIRT1, also plays a significant role in responding to CR. For example, CR elevates SIRT1 expression in a number of tissues [11], and transgenic mice that overexpress SIRT1 exhibit a phenotype mirroring some aspects of CR [12]. SIRT1 has also been shown to improve insulin sensitivity [13], another consequence of a CR diet [14].
CR promotes a healthy aging phenotype through a myriad of mechanisms, one of which is thought to be its ability to increase mitochondrial efficiency and biogenesis. Increases in mitochondrial biogenesis are driven by eNOS and PGC-1α expression and activation. Furthermore, these changes in mitochondria following CR are accompanied by a decrease in production of reactive oxygen species (ROS) without a net reduction of ATP biosynthesis, which indicates a higher bioenergetics efficiency [15,16]. There are several reports on how CR induces the deacetylation of PGC-1α by SIRT1[17]. Sirtuins are NAD+-dependent deacetylases [18], and this dependence has led researchers to propose that sirtuins are at the center of the regulatory nexus between energy metabolism and aging because NAD+ is a primary marker for intracellular energy status. It has also been demonstrated that CR activates sirtuins and thereby increases both the stability of chromatin [19] and cell survival [11]. Given the dependence of sirtuins on NAD+ and the published activities of sirtuins under CR conditions, it has been hypothesized that NAD+ levels and its metabolism are at the center of the regulatory mechanisms behind the beneficial effects of CR [17]. Furthermore, the conversion of NADH to its reduced form NAD+ in mitochondria, a reaction that is supported by coenzyme Q (CoQ), is also thought to protect mitochondria during aging [20]. We propose, therefore, mechanisms that affect the NAD+/NADH ratio and thereby modulate sirtuins and other NAD+-dependent enzymes are key players in the regulation of the aging process.
We have recently described the role of NQR1, a gene that encodes cytochrome b5 reductase, a protein that uses both NADH and CoQ as substrates, in chronological and replicative lifespan in Saccharomyces cerevisiae [21]. This enzyme is located at the plasma membrane and is homologous to the mammalian enzyme encoded by CYB5R3, which can also be found in plasma membranes and uses exclusively NADH and CoQ as substrates [22]. This enzyme is a key component of the trans-plasma membrane redox system (PMRS). The PMRS provides both protection against extracellular oxidants [23] and prevention of apoptosis initiated by the activation of the neutral sphingo-myelinase at the plasma membrane [24]. CR induces the expression of NQR1 in yeast, increasing the cytosolic NADH oxidation rate [21]. Similarly, CR increases the presence of this enzyme in the plasma membranes of both the liver and brain of rats, improving the antioxidant protection of phospholipids in these membranes [25,26]. This antioxidant system is also activated in mitochondrial DNA-deficient (ρ°) mammalian cells [27,28], and in vitamin E-deficient rat livers [29]. In the case of mammalian ρ° cells, cell survival is dependent on the redox homeostasis maintained by NADH oxidation by the PMRS. As indicated above, the increase of aerobic metabolism induced by CR also requires the cytosolic cooperation of CYB5R3 to maintain the NAD+/NADH ratio. Thus, any intervention that induces membrane instability or alters respiratory metabolism will evoke the transcription of CYB5R3 and activation of its enzymatic product.
Similar to the case in mammals, yeast NQR1 is upregulated by CR in parallel with an activation of respiration. Given that the same conditions activate the CoQ biosynthesis pathway [30], this may indicate a connection between CoQ biosynthesis and respiration. Interestingly, over-expression of NQR1 in yeast requires respiration to maintain cell survival. The mitochondrial mutant strains ΔATP2 and ΔCOR1 cannot grow under anaerobic conditions when NQR1 is overexpressed. The ΔATP2strain has a defective ATP synthase complex and the ΔCOR1 strain is defective in the bc1 complex. Similar results are obtained when the ΔCOQ2 strain, in which the CoQ biosynthesis pathway is inoperable, is used to overexpress NQR1. However, the addition of external CoQ6 restored both respiration and growth in the latest strain. These results indicate that NQR1 effect acts through the respiratory metabolism in yeast [21].
Over-expression of NQR1 extends chronological lifespan in the absence of SIR2, perhaps acting through a pathway dependent on NAD+/NADH balance that requires respiration [31], but not SIR2 [32]. NQR1 over-expression also extends replicative lifespan in a SIR2-dependent manner that mimics CR[8]. NQR1 promotes oxygen consumption while inhibiting ethanol production and this shift occurs alongside an increase in respiratory chain enzyme activities. NQR1 thus causes a shift from fermentative to respiratory metabolism that may help explain its role in longevity. Yeast growing in low glucose (CR) media also shows the increase of both chronological and replicative lifespan through the activation of respiration [8,31].
We can hypothesize then that NQR1 in yeast and CYB5R3 in mammals play a regulatory role connecting aerobic metabolism and aging processes through their ability to alter the NAD+/NADH ratio. Cytosolic NAD+/NADH must be balanced with that of mitochondria. We expect that NQR1 would partially prevent the de novo biosynthesis of NAD+ most likely by increasing the recycling of the redox state of nucleotides and maintaining the availability of NAD+ to consumer enzymes.
It is assumed that sirtuins connect metabolism to aging because they use NAD+ as substrate [18]. This rationale can also be applied to CYB5R3 because the enzyme consumes NADH as an obligatory substrate. This enzyme would then be an essential component of the NAD+/NADH-dependent metabolic pathways in cooperation with the mitochondrial respiratory chain (Figure 1), which both contribute to the maintenance of the NAD+/NADH ratio and, as a consequence, regulate the function of sirtuins and other downstream NAD+ consumers. The NADH consumers and NAD+ consumers may participate in a regulatory loop, as a decrease of NAD+ availability will activate NAD+ biosynthesis as has been shown to occur under stress such as in nutrient-dependent survival mechanisms [33].
Mammalian CYB5R3 may also connect aerobic metabolism and aging. CYB5R3 encodes for a membrane-bound form of cytochrome b5reductase in somatic cells that is N-myristoylated and thereby anchored to the plasma membrane, mitochondrial outer membrane and endoplasmic reticulum. This isoform participates in cholesterol biosynthesis [34], fatty acid elongation and desaturation [35], P-450 mediated hydroxylation of drugs and steroid hormones [36] and the PMRS [22]. There is also a soluble isoform, which lacks the N-terminal binding domain and exists in the cytoplasm of erythrocytes where its main function is to reduce methaemoglobin [37]. Both isoforms come from alternative splicing of the same CYB5R3 gene.Deficiencies of cytochrome b5reductase cause recessive congenital methaemoglobulinemia (RCM), which presents with two distinct clinical forms. RCM type I is benign and limited to red blood cells. RCM type II is severe, affects all cells in the organism, and can lead to neurological dysfunction (for review see [38]).
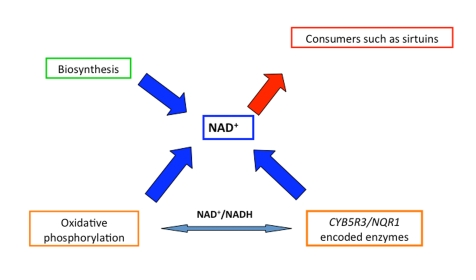
Figure 1. Role of the different characters to guarantee the availability of NAD+ to consumers maintaining at the same time the cellular redox homeostasis through a balanced NAD +/NADH ratio.
Recently, the proteomic profile of metabolic proteins in the invasive glioblastoma phenotype has been studied by applying a functional analysis using the Ingenuity Pathway Knowledge Base (Ingenuity Systems, Redwood City, CA) [39]. The results identified oxidative phosphorylation, mitochondrial dysfunction and ubiquinone biosynthesis as canonical pathways of the cancerous phenotype and CYB5R3 is identified as a protein associated with the mitochondrial dysfunction pathway. Furthermore, the relationship between mitochondrial dysfunction and CYB5R3 has also reported in a study carried out to analyze gene expression induced by bromide exposure using the Ingenuity Pathway Analysis[40].
Data from our laboratory seem to indicate a positive role for mammalian CYB5R3 in mitochondrial respiration. We have used siRNA technology to silence CYB5R3 in cultured human cells (Figure 2). Preliminary results indicate that CYB5R3 KO cells exhibit an apparent senescent phenotype based on the accumulation of β-galactosidase. These cells also show a reduction in the mitochondrial respiration rate based on analysis of oxygen consumption. Biochemical analysis of these cells also revealed an increase in the expression of PGC-1α that indicates increased recycling or de novo biogenesis of mitochondria. In a recently-reported global analysis of lysine-acetylated proteins, a posttranslational modifica-tion of CYB5R3-encoded protein by lysine acetylation in its FAD-binding domain has been identified [41]. Lysine acetylation is necessary for the interaction between SIRT1 and other sirtuins their targets before deacetylation can occur. Though conclusive experimental data still need to be shown, we hypothesize that SIRT1 regulates the cytosolic NAD+/NADH ratio by influencing CYB5R3 activity (Figure 3). Conditions of high NADH would lead to partial inactivation of SIRT1, leading to an accumulation of the acetylated form of CYB5R3. This active form of CYB5R3 would increase NADH oxidation and release NAD+ that, in turn, would activate SIRT1. CYB5R3 would be then deacetylated, causing a decrease in its activity and thereby maintaining the NAD+/NADH ratio in proper balance. PGC-1α activity will be also affected by this cycle through its interaction with SIRT1. Taken together, our preliminary data indicate CYB5R3 could play an essential role in the mitochondrial metabolism by its contribution to cellular redox homeostasis. A coordination of the redox balance in both the cytosol and mitochondria appears to be necessary for optimum cellular health, and may be of consequence to healthy aging as well.
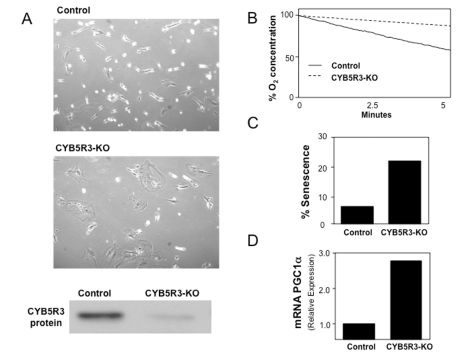
Figure 2. MRC-5 normal human diploid fibroblasts were CYB5R3-silenced (KO cells) and cultured in DMEM medium supplemented with FBS 10%. (A) Cell growth and CYB5R3 protein
levels after five days of CYB5R3-silencing are shown. (B)
Oxygen consumption was measured in parallel in both control and CYB5R3-KO
cells. (C) Percentage of senescence was determined by
senescence-associated-β-galactosidase activity. (D)
Total RNA was extracted in both control and CYB5R3-KO cells and PGC1α mRNA
levels were obtained by real time PCR.
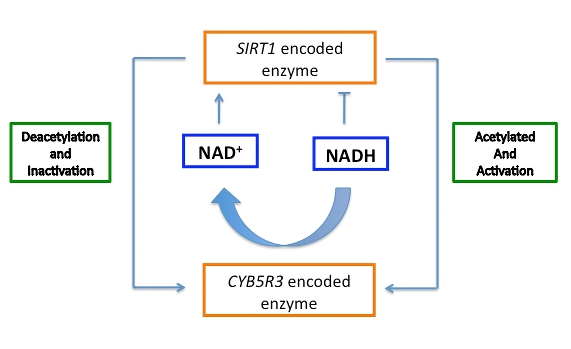
Figure 3. Hypothesis of the regulatory connection between cytochrome b5 reductase and sirtuin to maintain SIRT1 dependent respiration and cytosolic NAD+/NADH ratio.
Acknowledgments
This paper has been partially supported by Spanish FIS Grant PI080500, NIH Grant 1R01AG028125-01A1, and the Intramural Research Program of the NIH, National Institute on Aging.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Bartke A , Wright JC , Mattison JA , Ingram DK , Miller RA and Roth GS. Dietary restriction and life-span. Science. 2002; 296: 2141 -2142. [PubMed] .
- 2. Mattison JA , Roth GS , Lane MA and Ingram DK. Dietary restriction in aging nonhuman primates. Interdiscip Top Gerontol. 2007; 35: 137 -158. [PubMed] .
- 3. Weindruch R and Sohal RS. Seminars in medicine of the Beth Israel Deaconess Medical Center. Caloric intake and aging. N Engl J Med. 1997; 337: 986 -994. [PubMed] .
- 4. Canto C and Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009; 20: 325 -331. [PubMed] .
- 5. Lin SJ , Defossez PA and Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000; 289: 2126 -2128. [PubMed] .
- 6. Rogina B and Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004; 101: 15998 -16003. [PubMed] .
- 7. Chen J , Zhou Y , Mueller-Steiner S , Chen LF , Kwon H , Yi S , Mucke L and Gan L. SIRT1 protects against microglia-dependent beta amyloid toxicity through inhibiting NF-kappa B signaling. J Biol Chem. 2005; 280: 40364 -40374. [PubMed] .
- 8. Lin SJ , Kaeberlein M , Andalis AA , Sturtz LA , Defossez PA , Culotta VC , Fink GR and Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002; 418: 344 -348. [PubMed] .
- 9. Anderson RM , Bitterman KJ , Wood JG , Medvedik O and Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003; 423: 181 -185. [PubMed] .
- 10. Anderson RM , Latorre-Esteves M , Neves AR , Lavu S , Medvedik O , Taylor C , Howitz KT , Santos H and Sinclair DA. Yeast life-span extension by calorie restriction is independent of NAD fluctuation 1. Science. 2003; 302: 2124 -2126. [PubMed] .
- 11. Cohen HY , Miller C , Bitterman KJ , Wall NR , Hekking B , Kessler B , Howitz KT , Gorospe M , de Cabo R and Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004; 305: 390 -392. [PubMed] .
- 12. Bordone L and Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005; 6: 298 -305. [PubMed] .
- 13. Sung B , Park S , Yu BP and Chung HY. Modulation of PPAR in aging, inflammation, and calorie restriction. J Gerontol A Biol Sci. 2004; 59A: 997 -1006. .
- 14. Lane MA , Ingram DK and Roth GS. Calorie restriction in nonhuman primates: effects on diabetes and cardiovascular disease risk. Toxicol Sci. 1999; 52: 41 -48. [PubMed] .
- 15. Nisoli E , Tonello C , Cardile A , Cozzi V , Bracale R , Tedesco L , Falcone S , Valerio A , Cantoni O , Clementi E , Moncada S and Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005; 310: 314 -317. [PubMed] .
- 16. Lopez-Lluch G , Hunt N , Jones B , Zhu M , Jamieson H , Hilmer S , Cascajo MV , Allard J , Ingram DK , Navas P and de Cabo R. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006; 103: 1768 -1773. [PubMed] .
- 17. Canto C and Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009; 20: 98 -105. [PubMed] .
- 18. Finkel T , Deng CX and Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009; 460: 587 -591. [PubMed] .
- 19. Vaquero A and Reinberg D. Calorie restriction and the exercise of chromatin. Genes Dev. 2009; 23: 1849 -1869. [PubMed] .
- 20. Olgun A Converting NADH to NAD+ by nicotinamide nucleotide transhydrogenase as a novel strategy against mitochondrial pathologies during aging. Biogerontology. 2009; 10: 531 -534. [PubMed] .
- 21. Jimenez-Hidalgo M , Santos-Ocana C , Padilla S , Villalba JM , Lopez-Lluch G , Martin-Montalvo A , Minor RK , Sinclair DA , de Cabo R and Navas P. NQR1 controls lifespan by regulating the promotion of respiratory metabolism in yeast. Aging Cell. 2009; 8: 140 -151. [PubMed] .
- 22. Villalba JM , Navarro F , Cordoba F , Serrano A , Arroyo A , Crane FL and Navas P. Coenzyme Q reductase from liver plasma membrane: purification and role in trans-plasma-membrane electron transport. Proc Natl Acad Sci U S A. 1995; 92: 4887 -4891. [PubMed] .
- 23. Navas P , Villalba JM and de Cabo R. The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion 7 Suppl. 2007; 7: S34 -40. .
- 24. Villalba JM and Navas P. Plasma membrane redox system in the control of stress-induced apoptosis. Antioxid Redox Signal. 2000; 2: 213 -230. [PubMed] .
- 25. De Cabo R , Cabello R , Rios M , Lopez-Lluch G , Ingram DK , Lane MA and Navas P. Calorie restriction attenuates age-related alterations in the plasma membrane antioxidant system in rat liver. Exp Gerontol. 2004; 139: 297 -304. .
- 26. Hyun DH , Emerson SS , Jo DG , Mattson MP and de Cabo R. Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc Natl Acad Sci U S A. 2006; 103: 19908 -19912. [PubMed] .
- 27. Gomez-Diaz C , Villalba JM , Perez-Vicente R , Crane FL and Navas P. Ascorbate stabilization is stimulated in rho(0)HL-60 cells by CoQ10 increase at the plasma membrane. Biochem Biophys Res Commun. 1997; 234: 79 -81. [PubMed] .
- 28. Hyun DH , Hunt ND , Emerson SS , Hernandez JO , Mattson MP and de Cabo R. Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J Neurochem. 2007; 100: 1364 -1374. [PubMed] .
- 29. Navarro F , Navas P , Burgess JR , Bello RI , De Cabo R , Arroyo A and Villalba JM. Vitamin E and selenium deficiency induces expression of the ubiquinone-dependent antioxidant system at the plasma membrane. FASEB J. 1998; 12: 1665 -1673. [PubMed] .
- 30. Padilla S , Tran UC , Jimenez-Hidalgo M , Lopez-Martin JM , Martin-Montalvo A , Clarke CF , Navas P and Santos-Ocana C. Hydroxylation of demethoxy-Q6 constitutes a control point in yeast coenzyme Q6 biosynthesis. Cell Mol Life Sci. 2009; 66: 173 -186. [PubMed] .
- 31. Bonawitz ND , Chatenay-Lapointe M , Pan Y and Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007; 5: 265 -277. [PubMed] .
- 32. Smith DL Jr , McClure JM , Matecic M and Smith JS. Calorie restriction extends the chronological lifespan of Saccharomyces cerevisiae independently of the Sirtuins. Aging Cell. 2007; 6: 649 -662. [PubMed] .
- 33. Yang H , Yang T , Baur JA , Perez E , Matsui T , Carmona JJ , Lamming DW , Souza-Pinto NC , Bohr NA , Rosenzweig A , de Cabo R , Sauve AA and Sinclair DA. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007; 130: 1095 -1107. [PubMed] .
- 34. Reddy VV , Kupfer D and Caspi E. Mechanism of C-5 double bond introduction in the biosynthesis of cholesterol by rat liver microsomes. J Biol Chem. 1977; 252: 2797 -2801. [PubMed] .
- 35. Keyes SR and Cinti DL. Biochemical properties of cytochrome b5-dependent microsomal fatty acid elongation and identification of products. J Biol Chem. 1980; 255: 11357 -11364. [PubMed] .
- 36. Passon PG and Hultquist DE. Soluble cytochrome b5 reductase from human erythrocytes. Biochim Biophys Acta. 1972; 275: 62 -73. [PubMed] .
- 37. Jaffé ER Methemoglobin pathophysiology. Progress Clin Biol Res. 1981; 51: 133 -151. .
- 38. Percy MJ and Lappin TR. Recessive congenital methaemoglobinemia: cytochrome b5 reductase deficiency. Br J Haematol. 2008; 141: 298 -308. [PubMed] .
- 39. Rajcevic U , Petersen K , Knol JC , Loos M , Bougnaud S , Klychnikov O , Li KW , Pham TV , Wang J , Miletic H , Peng Z , Bjerkvig R , Jimenez CR and Niclou SP. iTRAQ-based proteomics profiling reveals increased metabolic activity and cellular cross-talk in angiogenic compared with invasive glioblastoma phenotype. Mol Cell Proteomics. 2009; 8: 2595 -2612. [PubMed] .
- 40. Price JA , Rogers JV , McDougal JN , Shaw MQ , Reid FM , Kiser RC and Graham JS. Gene expression analysis of bromine-induced burns in porcine skin. Toxicol Lett. 2008; 182: 69 -78. [PubMed] .
- 41. Choudhary C , Kumar C , Gnad F , Nielsen ML , Rehman M , Walther TC , Olsen JV and Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009; 325: 834 -840. [PubMed] .