




Introduction
As multicellular organisms age, somatic tissues show evidence of genomic instability and an increased error rate in protein synthesis. In contrast, the germ line is protected from genomic instability to ensure the ultimate survival of its genome. As the disposable soma theory of aging suggests, maintaining a low error rate is energy intensive, so somatic cells may trade off a high level of accuracy to save energy, leading to instability and eventually error catastrophe in aging somatic cells [1]. As embryonic stem cells (ESCs) can differentiate into all cell types, including the germ line, they must expend energy to maintain the genome and repair damage. Multiple stress defense mechanisms, such as telomere maintenance, antioxidant function, and DNA repair, are highly active in ESCs and downregulated during differentiation [2].
SIRT1 protects against age-related diseases by deacetylating targets (e.g., p53, FOXO, NFκB, and PGC-1α) that regulate diverse cellular processes, including stress response, replicative senescence, inflammation, and metabolism [3, 4]. SIRT1 protein levels are high in mouse embryonic stem cells [5, 6] and participates in the defense against oxidative stress in these cells [7]. Several Caenorhabditis elegans genes that ensure the genomic integrity of the germ line are also involved in regulating lifespan although it is not known if this protection is conserved in higher organisms [8]. As Sir2, the C. elegans homolog of SIRT1, regulates lifespan [9], SIRT1 may be a gene whose high-level expression in the germ line and ESCs maintains genomic integrity and plays a key role in regulating lifespan.
SIRT1 is critical for development: loss of both SIRT1 alleles in mice leads to postnatal lethality. Mice lacking SIRT1 survive when outbred but yield smaller, sterile mice with developmental defects [10, 11]. In addition, SIRT1 expression is induced during calorie restriction (CR), a 20-40% lowering of caloric intake that extends lifespan [12]. Transgenic mice that overexpress SIRT1 partially phenocopy CR [13], and are protected from age-related diseases such as diabetes, osteoporosis, and cancer [14]. SIRT1-/- mice do not have a longer lifespan on a CR diet [15]. Resveratrol, a polyphenol from grapes, works via the SIRT1 pathway to extend the lifespan of older mice fed a high-fat diet [16]. Similar to resveratrol, small-molecule activators of SIRT1 mimic the beneficial effects of CR and protect mice against age-related diseases [17, 18].
These observations highlight the importance of tightly regulating SIRT1 and the benefits of increasing SIRT1 expression and activity to promote longevity and suppress age-related diseases. Tight regulation of SIRT1 expression and activity is achieved through regulation of transcription by p53, FOXO3a, and E2F1 [19, 20]. SIRT1 expression is also regulated by controlling mRNA stability by HuR [21] and its enzymatic activity is sensitive to cellular NAD+ levels [22, 23] SIRT1-interacting proteins such as DBC1 and AROS also regulate its activity [24, 22].
Here we report that SIRT1 is highly expressed in mESCs compared to differentiated tissues and identify several miRNAs that regulate its expression at a post-transcriptional level during differentiation.
Results
SIRT1 protein is expressed at high levels in mESCs and post-transcriptionally downregulated during differentiation
We observed that SIRT1 protein levels are higher in mESCs than differentiated mouse tissues (Figure 1A). Overloading of lysate from differentiated tissues and a different SIRT1 antibody confirmed ubiquitous expression of SIRT1 in differentiated tissues, however expression was significantly lower levels than in mESCs (Figure 1A, lower panel). HDAC1 protein levels were also higher in mESCs, whereas HDAC2 protein expression was similar in mESCs and differentiated tissues (Figure 1A). Strikingly, measurement of SIRT1 mRNA levels by quantitative reverse transcription-PCR (qRT-PCR) showed relatively similar levels in mESCs and differentiated mouse tissues, except for skin and testis where mRNA levels were significantly higher (Figure 1B). In contrast, HDAC1 and HDAC2 mRNA correlated more closely with protein expression: HDAC1 mRNA levels were much lower (5-15 fold) in most differentiated tissues than in mESCs, whereas HDAC2 mRNA levels were similar in mESCs and differentiated tissues (Figure 1B). These findings of discordant mRNA and protein levels of SIRT1 suggested that SIRT1 is regulated post-transcriptionally in most adult mouse tissues.
To determine if SIRT1 is also regulated post-transcriptionally during in vitro differentiation of mESCs, we removed leukemia inhibitory factor (LIF) from the culture medium to allow the cells to differentiate into embryoid bodies. Protein and RNA were isolated from the mESCs and embryoid bodies every two days during in vitro differentiation. At d6 of the differentiation process, the high SIRT1 protein levels found in undifferentiated mESCs began to decrease (Figure 1C). Control mESCs cultured under non-differentiating conditions showed no change in SIRT1 expression (Figure 1C, right panel). In addition, SIRT1 protein expression levels decreased during directed differentiation of mESCs into neurons (Supplementary Figure 1). HDAC1 and HDAC4 expression were high in mESCs and decreased late during differentiation with kinetics distinct from that of SIRT1 (Figure 1C). In contrast, HDAC2 protein levels remained constant during in vitro differentiation. As expected, markers of pluripotency, including Nanog, Sox2, and Oct-3/4, were expressed in mESCs and decreased early during differentiation (Figure 1C and data not shown). In embryoid bodies, which exhibit spontaneous neural differentiation, the neuronal precursor marker Nestin was transiently induced, whereas Tau, a marker of mature neurons, was induced at late differentiation stages (Figure 1C).
In contrast to the decrease in SIRT1 protein levels observed during in vitro differentiation of mESCs, SIRT1 mRNA levels showed no change (Figure 1D, left panel). HDAC2 mRNA levels mirrored protein levels and were unchanged during differentiation. mRNAs levels of pluripotent stem cell markers, including Oct-3/4 (Figure 1D, left panel), Nanog, and Sox2 (data not shown) decreased during differentiation. mRNA expression of the ectoderm marker Map2 and the endoderm marker FoxA2 increased during differentiation, and Nestin mRNA expression transiently increased (Figure 1D, right panel).
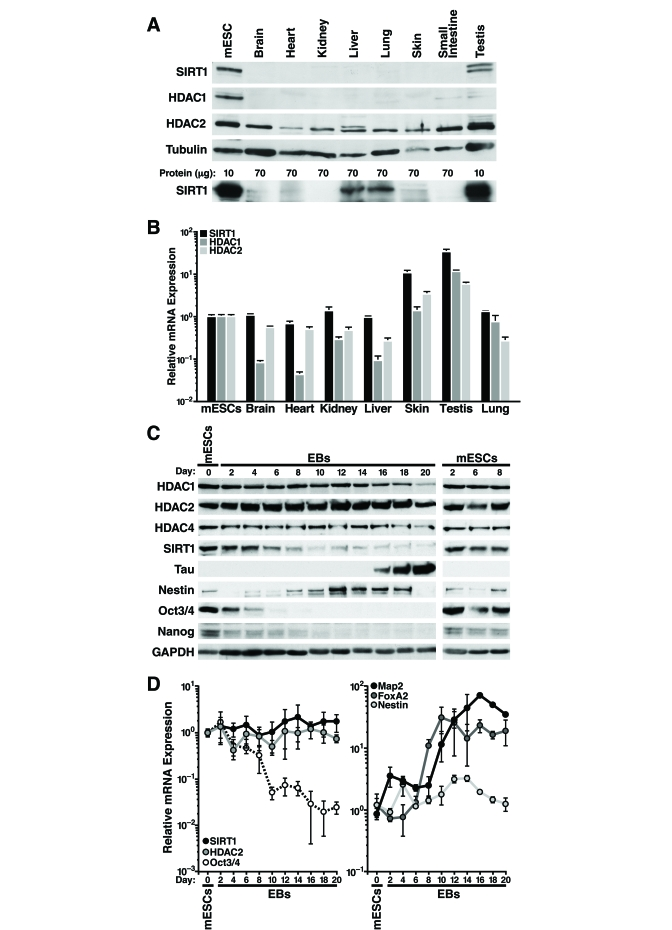
Figure 1. SIRT1 expression is regulated post-transcriptionally in adult mouse tissues and during mESC differentiation. (A-B) Protein and
RNA were extracted from mESC and tissues from ~6-week-old mice. (A)
Western blot analysis with antibodies against SIRT1 (Frye antiserum top
blot; Upstate antiserum lower blot), HDAC1, HDAC2, and tubulin. (B)
qRT-PCR analysis of SIRT1, HDAC1, and HDAC2 normalized to GAPDH levels.
Data are mean ± s.d. for four samples. (C-D) Protein and RNA were
isolated from mESCs differentiated in vitro for up to 20 days (EBs
d2-20). (C) Western blots analysis of expression of SIRT1, various
HDACs, markers of pluripotent embryonic stem cells, and markers of
differentiation. Data are representative of four experiments. (D)
qRT-PCR analysis of SIRT1, HDAC2, markers of pluripotent embryonic stem
cells, and markers of differentiation. Data were normalized to GAPDH and
plotted as expression relative to the mean of four
mESC samples. Data are mean
± s.d. for four samples.
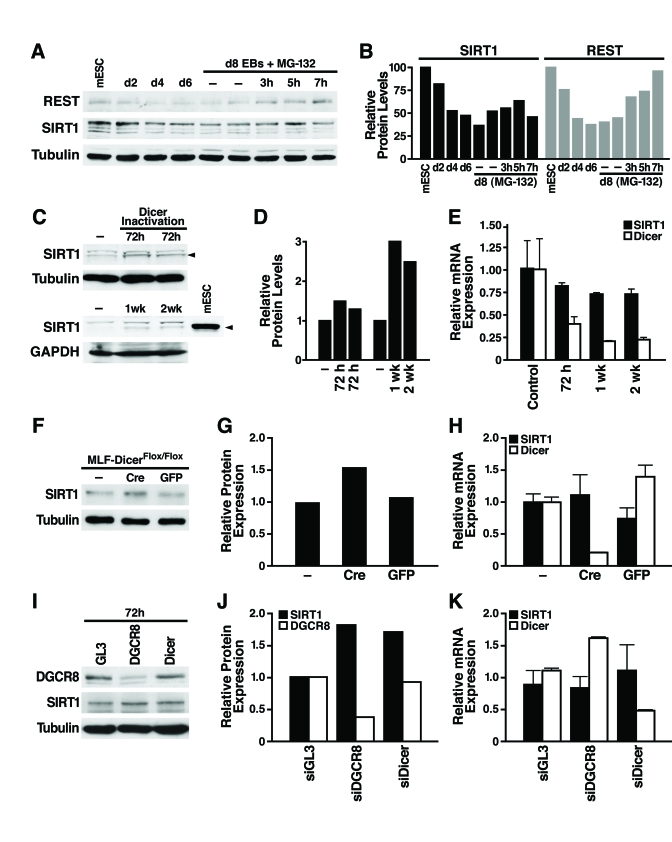
Figure 2. miRNAs post-transcriptionally regulate SIRT1. (A)
mESCs were differentiated and treated on d8 with the proteasome inhibitor
MG-132 (10 μM, 3-7 h), and
protein lysates were analyzed on western blots. Data are representative of
four experiments. (B) Protein levels of SIRT1 and REST relative to tubulin levels were quantified by densitometry
with NIH Image. (C-E) The
consequences of Dicer inactivation and loss of small RNAs were assessed in
protein lysates and RNA from livers of control and Dicerflox/flox
mice injected with the AAV8 vector expressing cre at the indicated times. (C)
Western blotting was used to analyze 70 μg
of liver lysate and 10 μg of mESC
lysate. (D) SIRT1 protein levels relative to tubulin or GAPDH were
quantified by densitometry. (E) SIRT1 and Dicer mRNA levels were measured
by qRT-PCR. Data are mean ± s.d. for four samples. (F-H) Lung
fibroblasts were cultured from DicerFlox/Flox mice and infected
with adenoviral Cre or GFP. (F) SIRT1 protein levels were measured
by western blotting 72 h after Cre inactivation of Dicer. (G) SIRT1
protein levels relative to tubulin were quantified by densitometry. (H)
mRNA levels of SIRT1 and Dicer were measured by qRT-PCR. Data are mean ±
s.d. for three samples. (I-K) siRNAs were transfected into NIH3T3
cells to knockdown DGCR8, Dicer, or GL3 luciferase as a control. (I)
DGCR8 knockdown and increased SIRT1 protein levels were analyzed by western
blotting 72 h after siRNA transfection. Data are representative of three
experiments. (G) qRT-PCR analysis confirmed Dicer knockdown and no
significant change in SIRT1 mRNA levels. Data are mean ± s.d. for three
samples.
A Dicer-dependent pathway post-transcriptionally regulates SIRT1 expression
To examine the mechanism of SIRT1 post-transcriptional regulation, we first tested whether SIRT1 protein stability is controlled by the proteasome. As a positive control, we confirmed that REST, an essential protein in undifferentiated mESCs that represses neuronal genes in differentiated non-neuronal tissues, was downregulated by the proteasome during differentiation as previously reported [26]. Treatment of d8 embryoid bodies with the proteasome inhibitor MG-132 increased REST protein expression; however, in the same cell culture population, proteasome inhibition did not increase SIRT1 protein expression (Figure 2A and B; Supplementary Figure 2). Thus, proteasome-mediated degradation of SIRT1 is not responsible for its post-transcriptional downregulation during differentia-tion.
We next determined if SIRT1 is subject to post-transcriptional regulation by miRNAs [27] For this purpose, we inactivated Dicer, an enzyme required for processing of small RNAs, including miRNAs, into their mature functional form [28] We injected Dicerflox/flox mice [29] with an adeno-associated viral (AAV) vector expressing Cre from the hepatocyte-specific transthyretin promoter. Liver-specific inactivation of Dicer increased SIRT1 protein levels (Figure 2C, D) while SIRT1 mRNA levels slightly decreased (Figure 2E). Additionally, we isolated lung fibroblasts from the Dicerflox/flox mice and infected them with an adenovirus expressing Cre or GFP. Cre-mediated inactivation of Dicer increased SIRT1 protein levels (Figure2F, G), without changing SIRT1 mRNA levels (Figure 2H). To determine whether miRNAs or other small RNAs regulate SIRT1 in differentiated tissues, we knocked down the expression of DGCR8, which is specifically required for processing of miRNAs, and Dicer in mouse NIH3T3 cells. Knockdown of either DGCR8 or Dicer increased SIRT1 protein expression (Figure 2I, J) without changing SIRT1 mRNA levels (Figure 2K). Knockdown of Dicer was verified by qRT-PCR mRNA measurement and knockdown of DGCR8 was verified by western blot (Figure 2 I-K). Thus, miRNAs post-transcriptionally regulate SIRT1 in differentiated tissues and cell lines, and may account for the downregulation of SIRT1 during in vitro mESC differentiation.
The SIRT1 mRNA 3'-UTR is targeted by multiple miRNAs
To identify miRNAs that target SIRT1, we examined the 1.6-kb mSIRT1 3'-UTR with algorithms that predict miRNA target sites [30, 31]. Target Scan 5.1 revealed 22 miRNAs targeting 12 broadly conserved seed sites in the 3'-UTR of mSIRT1. This analysis also revealed two miRNAs targeting three seed sites conserved only in mammals, and 66 seed sites for poorly conserved miRNA families. In contrast, HDAC1, which has a shorter 3'-UTR (0.5 kb), had no broadly conserved miRNA seed sites, one seed site conserved in mammals, and 22 seed sites for poorly conserved miRNAs (data not shown). We hypothesized that if miRNAs post-transcriptionally downregulate SIRT1 during mESC differentiation, the miRNAs responsible should be induced during differentiation when SIRT1 protein levels are decreased. We used qRT-PCR to profile the expression of 39 miRNAs that potentially target SIRT1: 21 well-conserved miRNAs (representing 11 miRNA families), two miRNAs conserved only in mammals, and 16 less conserved miRNAs many of which had two target sites in the 3'-UTR of mSIRT1 (Supplementary Table 1). We found that 18 miRNAs from nine families were upregulated 30-5000 fold during mESC differentiation (Figure 3A). The expression of six selected miRNAs during mESC differentiation is illustrated in Figure 3B,C.
miR-181a and b, miR-9, miR-204, miR-135a, and miR-199b target endogenous SIRT1
To identify miRNAs that post-transcriptionally regulate SIRT1, we cloned the 1.6-kb mSIRT1 3'-UTR downstream of luciferase, and transfected this construct (pGL3-SIRT1 3'-UTR) into mESCs along with miRNA mimics or miRNA expression constructs, and measured luciferase activity 24 h later. We found that miR-181a, b, and c repressed luciferase activity by 25-30% (Figure 4A, left panel). The specificity of this inhibition was demonstrated by testing the effect of the same miRNAs on a construct in which the miR-181 seed-binding site was mutated (pGL3-SIRT1 3'-UTR 181mt; Figure 4A, left panel). Likewise, co-transfection of a miR-9 expression vector repressed luciferase activity of pGL3-SIRT1 3'-UTR by 30% but not pGL3-SIRT1 3'-UTR 9mt, a control construct with a mutated miR-9 binding site (Figure 4A, right panel). Thus, miR-181 family members and miR-9 target the 3'-UTR of SIRT1 through the predicted seed sites.
To directly confirm the ability of select miRNAs to target the 3'-UTR of endogenous SIRT1, candidate miRNAs were introduced into mESCs and SIRT1 protein levels were assessed. Overexpression of miR-181a and b, miR-9, miR-204, miR-135a, and miR-199b decreased SIRT1 protein levels in mESCs (Figure 4B). In contrast, overexpression of miR-1, a miRNA not predicted to target the SIRT1 3'-UTR, did not decrease SIRT1 protein levels (Figure 4B). SIRT1 mRNA levels did not change upon miRNA overexpression, and the expression of individual miRNAs did not alter expression of other miRNAs (Figure 4C). These data confirm that miR-181a and b, miR-9, miR-204, miR-135a, and miR-199b target endogenous SIRT1 and downregulate its expression.
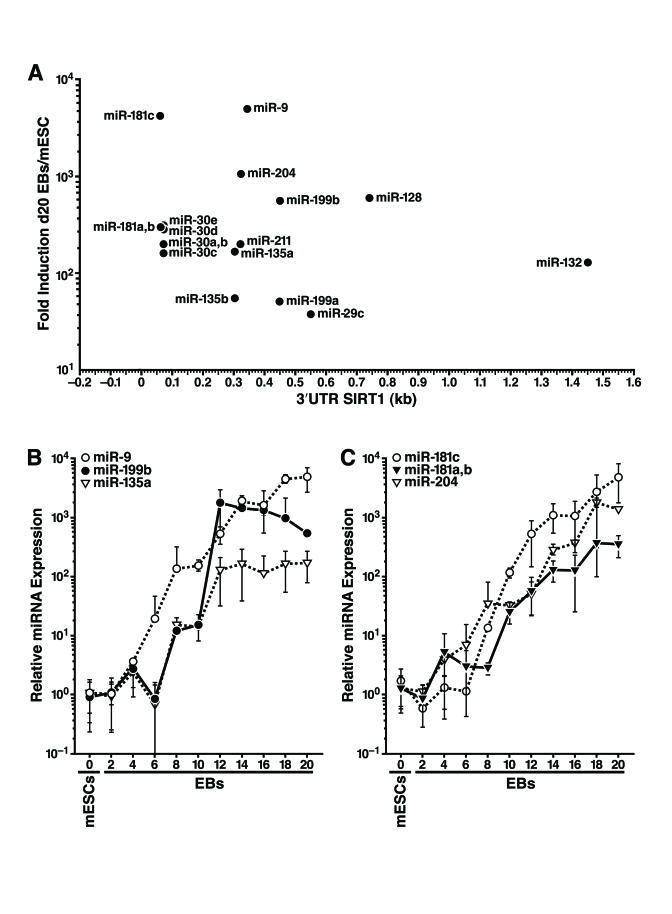
Figure 3. Expression profiling of miRNAs that potentially target the SIRT1 3'-UTR during mESC differentiation. (A) 18 miRNAs from nine miRNA
families that potentially target the 3'-UTR of SIRT1 were induced during
mESC differentiation at the time SIRT1 protein was downregulated. Their
fold induction in d20 embryoid bodies above their expression in undifferentiated
mESCs was plotted on the y-axis, and the location of their seed binding
site in the 3'-UTR of mSIRT1 was plotted on the x-axis. (B-C), qRT-PCR of
miRNA expression relative to miR-16 from undifferentiated mESCs and
differentiating embryoid bodies of specific miRNAs that potentially target
SIRT1. Data are mean ± s.d. for four samples.
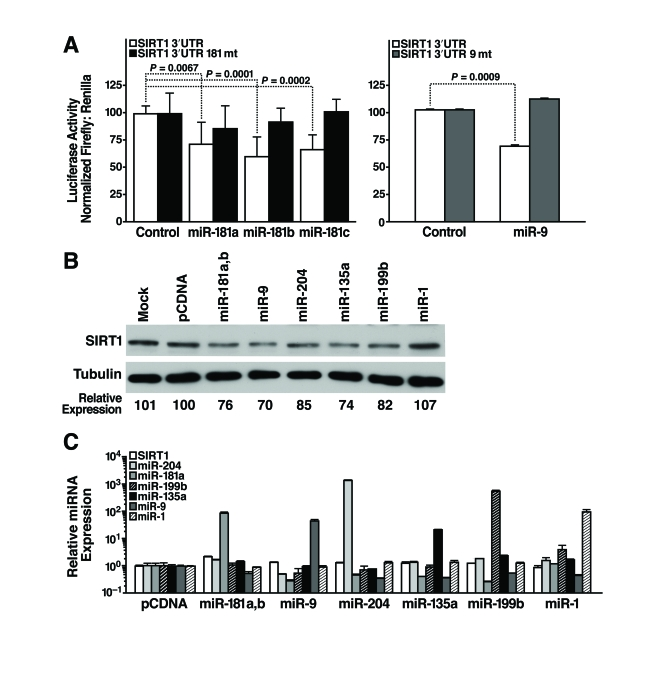
Figure 4. miRNAs post-transcriptionally regulate the 3'-UTR of SIRT1 mRNA. (A)
Luciferase assays were performed 24 h after transfection of the full-length
1.6 kb SIRT1 3'-UTR downstream of luciferase (SIRT1 3'-UTR)
or constructs with 4 bp in the seed-binding regions mutated (SIRT1 3'-UTR
181mt, left panel; SIRT1 3'-UTR 9mt, right panel) and control,
miR-181a, b, and c miRNA mimics (left panel) or pSuper and pSuper miR-9
expression constructs (right panel). Data are mean ± s.d. for eight
experiments. (B-C) mESCs were
transfected with individual miRNA expression constructs; protein and RNA
were isolated 48 h later. (B) Repression of SIRT1 protein was
analyzed by western blotting. Data are representative of six experiments. (C)
qRT-PCR analysis of SIRT1 mRNA levels and mature miRNA levels. Data are
mean ± s.d. for four samples.
Inhibition of miR-9 prevents the downregulation of SIRT1 protein expression during differentiation
We consistently observed that miR-9 was the first SIRT1-targeting miRNA to be upregulated both during differentiation of mESCs into embryoid bodies (Figure 3B) and during the directed differentiation of mESCs into neurons (data not shown). miR-9 is expressed in the brain, induced during differentiation of neuronal precursors into neurons, and regulates neural lineage differentiation [32]. To confirm that miR-9 represses SIRT1 early during mESC differentiation, we tested whether inhibition of miR-9 prevents the downregulation of SIRT1 protein. We used a FITC-labelled locked nucleic acid (LNA)-probe antisense to miR-9 to block miR-9 activity (LNA-miR-9). LNA-miR-9 or a scrambled control (LNA-SCR) was transfected into embryoid bodies at d4 and d7. Only cells on the outer layer of the embryoid bodies were transfected by this method, and fluorescence microscopy estimated that ~35% of cells were FITC+ (data not shown). As expected, miR-9 expression strongly increased during differentiation (Figure 5A). LNA-miR-9 reduced expression of miR-9 by 35% atday 8, but LNA-SCR did not. Neither inhibitor significantly altered SIRT1 mRNA expression (Figure 5B). Importantly, LNA-miR-9, but not LNA-SCR or untransfected controls, specifically prevented the differentiation-associated repression of SIRT1 protein (Figure 5C). Thus, of the 17 miRNAs upregulated during mESC differentiation that potentially target SIRT1, miR-9 acts early during differentiation to downregulate SIRT1 expression.
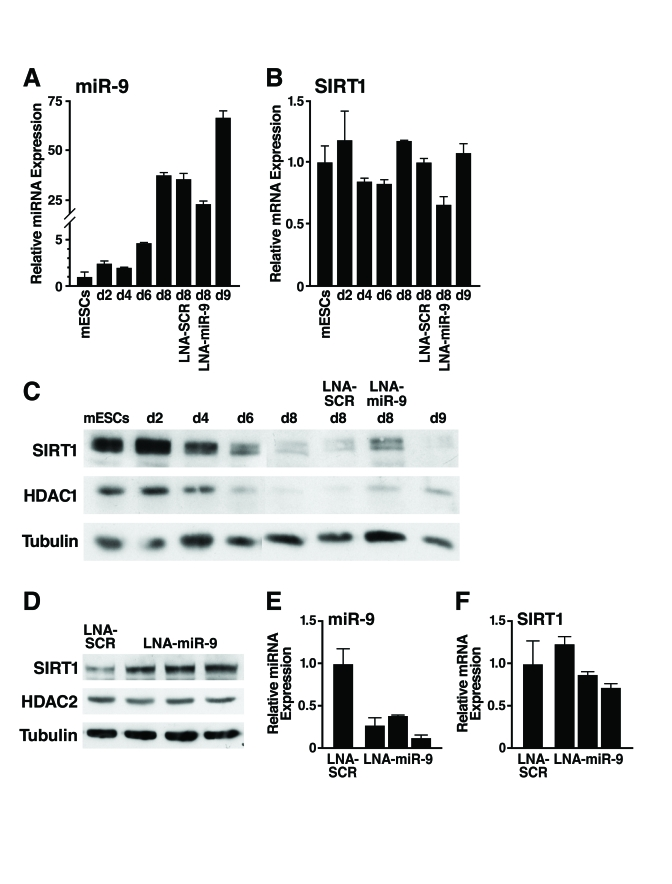
Figure 5. Inhibition of miR-9 prevents downregulation of SIRT1 during mESC differentiation.
(A-C) mESCs were
differentiated and transfected at d4 and d7 with LNA probes. Protein and
RNA were isolated on indicated days. (A) qRT-PCR of miR-9 shows the
expected upregulation during differentiation and 35% inhibition when
embryoid bodies were transfected with LNA-miR-9 but not with LNA-SCR. (B)
qRT-PCR show no significant change in SIRT1 mRNA levels. Data are mean ±
s.d. for four samples and representative of three experiments. (C)
Western blot analysis shows that the downregulation of SIRT1 protein during
mESC differentiation was specifically inhibited in cells transfected with
LNA-miR-9 but not by transfection of LNA-SCR or untransfected controls. Data
are representative of four experiments. (D-F) EBs were
dissociated and transfected at d6 with LNA probes. Protein and RNA were
isolated on d11. (D) Western blot analysis shows upregulation of
SIRT1 protein in EBs transfected with LNA-miR-9 but not LNA-SCR. (E)
qRT-PCR analysis shows inhibition of miR-9 in EBs transfected with
LNA-miR-9, but not with LNA-SCR, and no significant change in SIRT1 mRNA
levels (F). Data are mean ± s.d. for four samples.
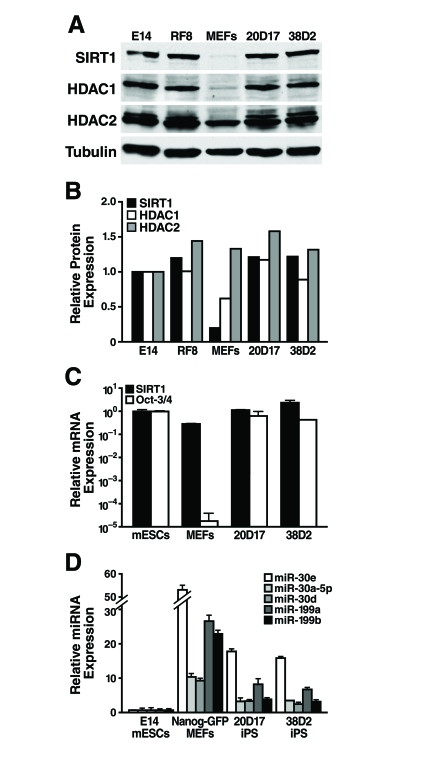
Figure 6. SIRT1 protein levels are upregulated during reprogramming. (A)
mESCs, MEFs, and iPS cells were subject to western blot analysis with
antibodies to the indicated proteins. (B) SIRT1,
HDAC1, and HDAC2 protein levels relative to tubulin were quantified by
densitometry. (C) qRT-PCR analysis of SIRT1 mRNA levels in mESCs,
MEFs, and iPS cells were measured relative to GAPDH. (D) qRT-PCR
analysis of miRNA expression relative to miR-16 in mESCs, MEFs, and iPS.
Data are mean ± s.d. for three samples.
To enhance the fraction of cells transfected, we dissociated d6 embryoid bodies, transfected them with LNA-miR-9 or LNA-SCR, reaggregated the embryoid bodies, and assessed SIRT1 expression at d11. With this method, 70-80% of the cells in the embryoid bodies were transfected, and LNA-miR-9 specifically increased SIRT1 protein levels ~two-fold (Figure 5D) qRT-PCR analysis demonstrated a more efficient repression of miR-9 expression in the LNA-miR-9 treated cells (Figure 5E), with minimal change in SIRT1 mRNA levels (Figure 5F). These observations confirmed that miR-9 inhibition increased SIRT1 protein levels.
SIRT1 protein levels increase during reprogramming
As SIRT1 protein levels are lower in differentiated tissues than in mESCs, we next asked if SIRT1 protein levels increase during reprogramming of mouse embryonic fibroblasts (MEFs) into induced pluripotent stem (iPS) cells. We used previously described iPS cell lines derived from retroviral mediated expression of Oct-3/4, Sox2, Klf4, and c-myc in MEFs. These iPS cell lines also express a Nanog-GFP reporter [33]. Protein levels of SIRT1 were low in the starting MEFs and were dramatically upregulated in iPS clones, to the same levels seen in two mESC lines, E14 and RF8 (Figure 6A, B). Similarly, low levels of HDAC1 protein were upregulated during reprogramming of MEFs into iPS, while HDAC2 protein levels were broadly similar in MEFs, iPS, and mESCs (Figure 6A, B). Comparison of SIRT1 mRNA levels in mESCs, MEFs, and iPS clones showed that the starting MEFs had only 30% of the SIRT1 mRNA, but this only partially explains the 6.5-fold difference in SIRT1 protein expression (Figure 6C). Thus, post-transcriptional regulation of SIRT1 contributes significantly to the upregulation of SIRT1 protein levels during reprogramming.
To identify miRNAs that may post-transcriptionally upregulate SIRT1 protein during reprogramming, expression levels of miRNAs that potentially target SIRT1 were compared in mESCs, MEFs, and iPS cells. As previously discussed, miR-199a and b were strongly upregulated during mESC differentiation (Figure 3). As predicted, reprogramming of MEFs into iPS cells was accompanied by a downregulation of miR-199a and b by 3.3-fold and 5.8-fold, respectively (Figure 6D). Additionally, all five members of the miR-30 family that potentially target SIRT1 were higher in MEFs than iPS and mESCs. Therefore, expression of select miRNAs, including the miR-199 and miR-30 families, decreases during reprogramming and may allow for the upregulation of SIRT1 protein expression.
Discussion
Our work shows that SIRT1 is highly expressed in mESCs and that miRNAs post-transcriptionally downregulate SIRT1 protein expression during mESC differentiation and maintain low SIRT1 protein levels in differentiated adult mouse tissues. Specifically, SIRT1 expression is repressed by miR-181a and b, miR-9, miR-204, miR-135a, and miR-199b.
Repression of SIRT1 protein expression by miRNAs may play an important role in development since several miRNAs that target SIRT1 have previously been identified as regulators of specific differentiation pathways. For example, miR-9, a miRNA expressed early during mESC differentiation, participates in neuronal differentiation [32]. Since activation of SIRT1 in neuronal precursors promotes astrocyte formation over neurogenesis [34], SIRT1 might represent a critical target for miR-9. Another similar example is miR-181, which is transiently upregulated during muscle differentiation [35]. SIRT1 inhibition induces premature differentiation of C2C12 myoblasts, and SIRT1 activation inhibits muscle differentiation [36]. Thus, regulation of SIRT1 by miR-181 might contribute to the muscle differentiation program. miR-181a also regulates T-cell-receptor sensitivity and signal strength during T-cell development, in part by targeting tyrosine phosphatases [37]. Since SIRT1 inhibition induces T-cell hyperactivation [38], miR-181a may also target SIRT1 during T cell development.
Because each miRNA targets only one site in the SIRT1 3'-UTR, multiple tissue-specific miRNAs likely work together to regulate SIRT1 expression. Additionally, miRNA regulation of SIRT1 might be influenced by HuR [39], which binds the 3'-UTR and stabilizes the SIRT1 transcript [21], even though HuR binding sites do not directly overlap miRNA seed-binding sites in the SIRT1 3'-UTR. HuR, whose expression decreases during aging, is targeted by miR-519, which triggers senescence and represses tumor growth through downregulation of HuR [40, 41]. Tissue-specific therapeutic targeting of miRNAs that regulate SIRT1 might allow the selective upregulation of SIRT1 in unique tissues, whereas current small molecules that activate SIRT1 do so in a tissue non-specific manner.
We also tested whether SIRT1 protein levels might increase upon reprogramming of MEFs into iPS cells. Remarkably, we found that low SIRT1 protein levels in MEFs were upregulated during reprogramming into iPS cells to levels similar to mESCs (Figure 6A). This correlated with the downregulation of miR-199 andmiR-30 families that target SIRT1 (Figure 6C). Expression of miR-199a and b is highest in skin (Supplementary Figure 3), and limiting the expression of these specific miRNAs may be a prerequisite for reprogramming of MEFs. Reprogramming of other differentiated cell types may require downregulation of distinct tissue-specific miRNAs that regulate SIRT1 expression.
An important area for future focus will be to understand why SIRT1 protein levels are exceptionally high in mESCs. SIRT1 might be required to maintain a unique chromatin state in ESCs, or to deacetylate non-histone targets that are essential for early development. For example, SIRT1 deacetylates HSF1 to enhance its activity [42], and maternal HSF1 is required for development beyond the zygote stage [43]. Therefore, high expression of SIRT1 may work together with HSF1 during early development.
However, SIRT1 is not absolutely required during early development. Loss of SIRT1 on an outbred genetic background allows for 50% of SIRT1-/- mice to develop relatively normally [11]. Importantly, other SIRT1 null mouse models show that SIRT1-/- mice are not obtained at expected ratios with the majority of SIRT1-/- mice dieing right after birth [10] or between E9.5 and E14.5 [44]. It is possible that another deacetylase, namely HDAC1, which is also both highly expressed in mESCs (Figure 1A) and upregulated during reprogramming (Figure 6A), partially compensates for SIRT1. In support of this idea, many non-histone targets are deacetylated by both SIRT1 and HDAC1 including p53 and NF-κB [4].
At least in lower organisms, SIRT1 regulates lifespan, and several genes that regulate lifespan also maintain genomic integrity in germ cells and stem cells [8]. A possible role of SIRT1 in mESCs and during early development could be to monitor quality control of developing embryos. SIRT1 may respond to oxidative stress, genotoxic damage, metabolic defects, and epigenetic reprogramming errors, possibly through the deacetylation of p53 and other targets, to regulate survival of developing embryos. Indeed, expression level and activity of p53 in early pre-implantation embryos regulates their viability [45].
Another intriguing question is whether downregulation of SIRT1 is necessary during differentiation and development. SIRT1 may be downregulated during differentiation in a manner similar to other stress defense mechanisms that are highly active in ESCs [2]. The downregulation of SIRT1 via a post-transcriptional mechanism allows its mRNA to persist and might allow SIRT1 expression to be rapidly induced during stress when energy intensive cell repair and survival mechanisms are required. The decrease of SIRT1 protein levels observed during aging may conserve energy but may also contribute to increased genomic instability [46].
Loss of miRNAs might contribute to the overexpression of SIRT1 in cancer. For example, loss of miR-34a leads to SIRT1 overexpression in cancer [47, 48]. Some results point to direct binding of miR-34a to the SIRT1 3'-UTR whereas others have suggested indirect regulation of SIRT1 by miR-34a [47, 48]. Several other miRNAs that target SIRT1 are lost in cancers. For example, miR-181a and b function as tumor suppressors in the brain, but their loss negatively correlates with glioma grade, and restoration of their expression induces apoptosis of glioma cells [49]. Furthermore, miR-181 and miR-29 family members are downregulated in chronic lymphocytic leukemia, and miR-29 is lost in colon, breast, and lung cancer [50, 53]. While SIRT1 may function as a tumor suppressor by limiting replicative senescence in primary cells, SIRT1 overexpression is seen in many cancers where it may promote cell survival [4]. Reintroduction of miRNAs lost in cancers that overexpress SIRT1 may be of therapeutic value against cancers dependent on the overexpression of SIRT1.
Our findings that miRNAs regulate SIRT1 expression suggest that inhibiting specific miRNAs may be of therapeutic value in disease conditions where SIRT1 activity has been shown to be beneficial such as diabetes, neurodegeneration, and cancer [54]. Currently available small molecule SIRT1 activators and inhibitors globally increase or inhibit SIRT1 activity. In contrast, the use of tissue-specific miRNA mimics or inhibitors may allow for the tissue-specific regulation of SIRT1 to prevent and treat age-related diseases without globally altering SIRT1 activity.
Methods
Culturing and differentiation of mESCs. E14 mESCs [55] were cultured feeder-free in Glasgow MEM/BHK12 (GMEM; Sigma-Aldrich; St. Louis, MO) supplemented with 10% characterized fetal bovine serum (FBS; Hyclone; Logan, UT), 2 mM L-glutamine (GIBCO Invitrogen Corporation; Carlsbad, CA), 1 mM sodium pyruvate (GIBCO Invitrogen Coroporation), 0.5 mM β-mercaptoethanol (Sigma), and leukaemia inhibitory factor (LIF) conditioned medium on plates coated with 0.1% bovine gelatin (Sigma) in PBS. Undifferentiated ESCs were passaged every 2 days, and medium was changed on alternate days. Differentiation was induced by plating 3x106 cells in 10-cm, ultra-low attachment dishes (Corning; Lowell, MA) in 10 ml of differentiation medium (GMEM supplemented with 15% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, and 0.5 mM β-mercaptoethanol). Medium on the embryoid bodies was changed every 2 days. The proteasome inhibitor MG-132 (10 μM; Calbiochem; Darmstadt, Germany) was added to the media of d8 embryoid bodies for the indicated times.
For neuronal differentiation, 5 μM retinoic acid (Sigma R-2625) was added to d4 embryoid bodies [56]; then d8 embryoid bodies were trypsinized to form a single-cell suspension. Cells were strained through a 40-μM nylon mesh (BD Biosciences; San Jose, CA), and 8x105 cells in 1 ml of neurobasal A (NBA) medium (Invitrogen) supplemented with 2% B27 supplement (Invitrogen) and 500 μM glutamine were plated onto poly-D-lysine/mouse laminin 12-mm coverslips (BD Biosciences) in 24-well plates. Medium was changed 2 and 24 h after plating. After 2 days, the medium was changed to NBA supplemented with 1% N2 (Invitrogen) and 500 μM glutamine.
Expression constructs. The full-length 1.6 kb mSIRT1 3'-UTR was PCRed from IMAGE clone 3587177 (Open Biosystems; Huntsville, AB) with primers that add NheI sites (underlined): forward, 5'-TCATAACGCTAGC GA AGCTGTCCG-3'; reverse, 5'-TCCAGTCATTAAACG GGCTAGC AAAC-3'. This SIRT1 3'-UTR was cloned behind luciferase in the pGL3-promoter vector (Promega; Madison, WI) digested with XbaI. Site-directed mutagenesis was performed using a QuikChange II Site-Directed Mutagenesis kit (Stratagene; La Jolla, CA) to mutate base pairs 3-6 in the predicted seed region targeted by miR-181 and miR-9 in the SIRT1 3'-UTR.
Genomic DNA 250-350 bp on either side of the genomic locus for miR-181a and b, miR-9, miR-204, miR-135a, and miR-199b was amplified and cloned into pCDNA/V5-DEST (Invitrogen) with the following primers: mmu-miR-181a and mmu-miR-181b amplified from chromosome 1 (5'-CACCAACAGCCTGTAACT AAGCTCC-3' and 5'-TGATTCTGGGCATCCAACAC -3'), mmu-miR-9-2 amplified from chromosome 13 (5'-CTAGCCGCACACACTAAG-3' and 5'-TGCATCCCA CTTTCAATCATA-3'), mmu-miR-204 amplified from chromosome 19 (5'-CACCTTCATTCAGCACCTAGT TGAG-3' and 5'-ATACATTACAACCTGTTCAGAGG -3'), mmu-miR-199b amplified from chromosome 2 (5'-CCACAGGAGGCAGAAGGGGAGTCG-3' and 5'-CCCATCAGCCCAGCCATTTGC-3'), and mmu-miR-135a amplified from chromosome 9 (5'-CACCTCAG TGTCCAATGGGAATAC-3' and 5'-GGCTATCAAGG GGTTTCTTCAGG-3'). miR-1 was cloned as described [57].
Western blot analysis . mESCs, Embryoid bodies, and neurons were lysed in 50 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, 150 mM NaCl, 0.5% NP-40, and 1x complete protease inhibitors (Roche; Penzberg, Germany), and protein concentrations were determined with the DC Protein Assay (Bio-Rad). Organs harvested from ~6-week-old mice were lysed (0.1 g/ml) in 50 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, 500 mM NaCl, 0.5% NP-40, and 1x complete protease inhibitors (Roche) with a Dounce homogenizer. Protein samples were separated by electrophoresis on 7.5% or 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked with 5% nonfat dry milk in TBS-Tween [10 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.1% Tween-20] and probed with antiserum against HDAC1 [58], HDAC2 (Santa Cruz #7899), SIRT1 (polyclonal antiserum to amino acids 506-747 of hSIRT1 or Millipore #07-131), GAPDH (Novus Biologicals; Littleton, CO), Actin (Sigma), HDAC4 [59], Tau (EMD Biosciences; Germany), Nestin (Millipore; Billerica, MA), Oct-3/4 (R&D Systems), Nanog (Cosmo Bio; Tokyo, Japan), REST (Millipore), DGCR8 (Proteintech; Chicago, IL) and α-tubulin (Sigma).
Quantitative RT-PCR. Total RNA was isolated using TRIzol (Invitrogen). With Superscript II or III (Invitrogen) and oligo dT, 1 μg of RNA was reverse transcribed into cDNA. with Superscript II or III (Invitrogen) and oligo dT. Relative expression levels were determined by real-time quantitative PCR in an ABI 7700 or 7900 and normalized to GAPDH. 2X HotSybr Real-time PCR mix (McLab; South San Francisco, CA) was used with validated primers for HDAC1 (PPM04372A), HDAC2 (PPM04361A), and SIRT1 (PPM05054A; SuperArray Bioscience; Frederick, MD). GAPDH was amplified using (forward: 5'-ACTCCACT CACGGCAAATTCA, reverse: 5'-GCCTCACCCCATT TGATGTT), Oct-3/4 was amplified using (forward: 5'- TCAGCCTTAAGAACATGTGTAAGC, reverse: 5'- GTCTCCGATTTGCATATT CTCC), and Dicer was amplified using (forward 5'- TGGGAGATGCGATTT TGGA, reverse: 5'- GCTGCCC GTGGGTCTTCATAA). 2X HoTaq Real-time PCR mix (McLab) was used with validated primers from Applied Biosystems for Nestin (Mm00450205_m1), SIRT1 (Mm_00490758_m1), FoxA2 (Mm01976556_ s1), and Map2 (Mm00485230_m1).
Relative miRNA expression levels were quantified using the NCode miRNA first-strand cDNA synthesis kit (Invitrogen) to add a polyA tail onto the miRNAs. qPCR was performed using a forward primer to the exact sequence of the target miRNA and a reverse primer provided in the NCode kit. cDNA and qPCR reactions were generated using validated primers (Applied Biosystems) for hsa-miR-16 (4373121), has-miR-181a (4373117), hsa-miR-9 (4373285), has-miR-204 (4373313), has-miR-199b (4373309), has-miR-135a (4373140), and hsa-miR-1 (4395333).
AAV8 vector preparation and Adenovirus adenovirus infection. The double-stranded AAV8 vector for the expression of Cre from the transthyretin promoter was described (Amar Deep Sharma et al., manuscript submitted). Briefly, A293 cells were transfected with the AAV vector plasmid, the adenoviral helper plasmid pAd5, and the AAV8 capsid expression plasmid p5E18-VD2/8 [60] by the calcium phosphate method. Virus was collected 72 h after transfection and concentrated by centrifugation on cesium chloride density gradients. Viral titer was determined by dot blot analysis. Viral particles (2 x 1011 in 100 μl) were injected into the tail vein of Dicerflox/flox mice [11]. Livers were harvested 72 h, 1 wk, and 2 wk after virus injection.
Lungs from Dicerflox/flox mice were cut into small pieces and adhered to tissue culture plates in DMEM. Fibroblasts that grew out of the explants were collected and 80,000 lung fibroblasts were seeded in 1ml of DMEM into a 12-well plate. 24 h later, adenovirus expressing GFP or Cre was added at an MOI=100 to 500 μl of fresh DMEM in each well. Protein and RNA were isolated 72 h later.
siRNAs, miRNA mimics, and LNA probes. 20,000 NIH3T3 cells were plated per well of a 12-well plate in 1 ml of DMEM with 10% bovine calf serum without antibiotics 24 h before transfection. siGENOME SMARTpool siRNAs (10 nM) against DGCR8, Dicer, or GL3 luciferase (Thermo Scientific) were added to 100 μl of OptiMem. Lipofectamine RNAiMax (2 μl) (Invitrogen) in 98 μl of OptiMem was mixed with the siRNA for 20 min. This 200-μl solution was added along with 800 μl of fresh medium to each well. Protein and RNA were isolated at indicated time points.
miRNA mimics in the form of siRNA duplexes (Thermo Fisher Scientific; Waltham, MA) for mmu-miR-181a (C-310047-04), mmu-miR-181b (C-310182-05), mmu-miR-181c (C-310183-02), the microRNA mimic negative control (CN-001000-01), and FITC-conjugated miRCURY LNA knockdown probes (Exiqon; Woburn, MA) antisense to mmu-miR-9 (LNA-miR-9; 139459-04) or scramble control (LNA-SCR; 199002-04) were transfected into mESCs or embryoid bodies using lipofectamine 2000 (Invitrogen). Embryoid bodies were transfected by trypsininzing embryoid bodies to single-cell suspensions. 700,000 cells in 600 μl of medium were added to complexes containing 4 μl of the 25 μM LNA probe and 5 μl Lipofectamine 2000 in 300 μl of OptiMem. The cells were plated in 24-well ultra-low-attachment plates, and after 30 min, 750 μl of medium was added.
mESCs (2.5x105 in 300 μl of medium) were added to complexes containing 1.6 μg of pCDNA miRNA expres-sion vectors and 3 μl of Lipofectamine 2000 (Invitrogen) in 150 μl OptiMem. The cells were plated on gelatinized 12-well plates, and 1.5 ml of medium was added after 30 min, and medium was changed the next day.
Luciferase assays. mESCs (150,000 in 1 ml of medium) were added to gelatinized 24-well plates and immediately transfected using 1 μl Lipofectamine 2000 (Invitrogen) with 20 ng Renilla luciferase as an internal control, 200 μg pGL3-SIRT1 3'-UTR or vectors with mutated seed sites, and 20 pmol (~300 ng) of the miRNA mimics or 200 ng of an miRNA expression construct. After 24 h, cells were washed in 1X PBS, lysed at room temperature for 15 min in 100 μl of 1X passive lysis buffer (Promega), and 20 μl of the lysate was used in a dual luciferase assay (Promega) in a Monolight 2010 luminometer (Analytical Luminescence Laboratory; San Diego, CA). Results were normalized to Renilla and are shown relative to samples cotransfected with a negative control miRNA or empty miRNA expression vector.
Supplementary Materials
SIRT1 protein is down-regulated during directed differentiation of mESCs into neurons. Western analysis of SIRT1, REST, HDAC2, Oct-3/4, and Tau during directed differentiation of mESCs into neurons by treatment with retinoic acid and plating on poly-D-lysine/laminin coated plates.
SIRT1 is not post-transcriptionally regulated by the proteasome during mESC differentiation. d8 EBs were treated for the indicated times with the proteasome inhibitor MG-132 and analyzed by western blotting for expression of REST, SIRT1, and tubulin.
miR-199 is highly expres-sed in mouse embryonic fibroblasts and skin. qRT-PCR analysis of miR-199a and b expression relative to miR-16 in mESCs, MEFs, and various mouse tissues. Data are mean ± s.d. for four samples.
miRNAs that potentially target SIRT1.
Acknowledgments
We thank Kathy Ivey for suggestions on the manuscript, Roy Frye for SIRT1 antiserum, Deepak Srivastava for miR-1 construct, the staff of the Gladstone Stem Cell Core for support, Gary Howard for editorial assistance, and John Carroll, Teresa Roberts, and Chris Goodfellow for graphics assistance. This research was supported by a fellowship (L.R.S.) from the California Institute of Regenerative Medicine.
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Kirkwood TB Evolution of ageing. Nature. 1977; 270: 301 -304. [PubMed] .
- 2. Saretzki G , Walter T , Atkinson S , Passos JF , Bareth B , Keith WN , Stewart R , Hoare S , Stojkovic M , Armstrong L , von Zglinicki T and Lako M. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells. 2008; 26: 455 -464. [PubMed] .
- 3. Longo VD and Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006; 126: 257 -268. [PubMed] .
- 4. Saunders LR and Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007; 26: 5489 -5504. [PubMed] .
- 5. McBurney MW , Yang X , Jardine K , Bieman M , Th'ng J and Lemieux M. The absence of SIR2alpha protein has no effect on global gene silencing in mouse embryonic stem cells. Mol Cancer Res. 2003; 1: 402 -409. [PubMed] .
- 6. Kuzmichev A , Margueron R , Vaquero A , Preissner TS , Scher M , Kirmizis A , Ouyang X , Brockdorff N , Abate-Shen C , Farnham P and Reinberg D. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc Natl Acad Sci U S A. 2005; 102: 1859 -1864. [PubMed] .
- 7. >Han MK , Song EK , Guo Y , Ou X , Mantel C and Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008; 2: 241 -251. [PubMed] .
- 8. Curran SP , Wu X , Riedel CG and Ruvkun G. A soma-to-germline transformation in long-lived Caenorhabditis elegans mutants. Nature. 2009; 459: 1079 -1084. [PubMed] .
- 9. Tissenbaum HA and Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001; 410: 227 -230. [PubMed] .
- 10. Cheng HL , Mostoslavsky R , Saito S , Manis JP , Gu Y , Patel P , Bronson R , Appella E , Alt FW and Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003; 100: 10794 -10799. [PubMed] .
- 11. McBurney MW , Yang X , Jardine K , Hixon M , Boekelheide K , Webb JR , Lansdorp PM and Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003; 23: 38 -54. [PubMed] .
- 12. Cohen HY , Miller C , Bitterman KJ , Wall NR , Hekking B , Kessler B , Howitz KT , Gorospe M , de Cabo R and Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004; 305: 390 -392. [PubMed] .
- 13. >Bordone L , Cohen D , Robinson A , Motta MC , van Veen E , Czopik A , Steele AD , Crowe H , Marmor S , Luo J , Gu W and Guarente L. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007; 6: 759 -767. [PubMed] .
- 14. Herranz D and Serrano M. Impact of Sirt1 on mammalian aging. Aging. 2010; 2: .
- 15. Boily G , Seifert EL , Bevilacqua L , He XH , Sabourin G , Estey C , Moffat C , Crawford S , Saliba S , Jardine K , Xuan J , Evans M , Harper ME and McBurney MW. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS ONE. 2008; 3: e1759 [PubMed] .
- 16. Baur JA , Pearson KJ , Price NL , Jamieson HA , Lerin C , Kalra A , Prabhu VV , Allard JS , Lopez-Lluch G , Lewis K , Pistell PJ , Poosala S , Becker KG , Boss O , Gwinn D , Wang M , Ramaswamy S , Fishbein KW , Spencer RG , Lakatta EG , Le Couteur D , Shaw RJ , Navas P , Puigserver P , Ingram DK , de Cabo R and Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006; 444: 337 -342. [PubMed] .
- 17. Milne JC , Lambert PD , Schenk S , Carney DP , Smith JJ , Gagne DJ , Jin L , Boss O , Perni RB , Vu CB , Bemis JE , Xie R , Disch JS , Ng PY , Nunes JJ , Lynch AV , Yang H , Galonek H , Israelian K , Choy W , Iffland A , Lavu S , Medvedik O , Sinclair DA , Olefsky JM , Jirousek MR , Elliott PJ and Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007; 450: 712 -716. [PubMed] .
- 18. Feige JN , Lagouge M , Canto C , Strehle A , Houten SM , Milne JC , Lambert PD , Mataki C , Elliott PJ and Auwerx J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008; 8: 347 -358. [PubMed] .
- 19. Nemoto S , Fergusson MM and Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004; 306: 2105 -2108. [PubMed] .
- 20. Wang C , Chen L , Hou X , Li Z , Kabra N , Ma Y , Nemoto S , Finkel T , Gu W , Cress WD and Chen J. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat Cell Biol. 2006; 8: 1025 -1031. [PubMed] .
- 21. Abdelmohsen K , Pullmann R Jr , Lal A , Kim HH , Galban S , Yang X , Blethrow JD , Walker M , Shubert J , Gillespie DA , Furneaux H and Gorospe M. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell. 2007; 25: 543 -557. [PubMed] .
- 22. Imai S , Armstrong CM , Kaeberlein M and Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000; 403: 795 -800. [PubMed] .
- 23. Landry J , Sutton A , Tafrov ST , Heller RC , Stebbins J , Pillus L and Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A. 2000; 97: 5807 -5811. [PubMed] .
- 24. Kim EJ , Kho JH , Kang MR and Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007; 28: 277 -290. [PubMed] .
- 25. Kim JE , Chen J and Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008; 451: 583 -586. [PubMed] .
- 26. Ballas N , Grunseich C , Lu DD , Speh JC and Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005; 121: 645 -657. [PubMed] .
- 27. Zhao Y and Srivastava D. A developmental view of microRNA function. Trends Biochem Sci. 2007; 32: 189 -197. [PubMed] .
- 28. Valencia-Sanchez MA , Liu J , Hannon GJ and Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006; 20: 515 -524. [PubMed] .
- 29. Harfe BD , McManus MT , Mansfield JH , Hornstein E and Tabin CJ. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci U S A. 2005; 102: 10898 -10903. [PubMed] .
- 30. Krek A , Grun D , Poy MN , Wolf R , Rosenberg L , Epstein EJ , MacMenamin P , da Piedade I , Gunsalus KC , Stoffel M and Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005; 37: 495 -500. [PubMed] .
- 31. Grimson A , Farh KK , Johnston WK , Garrett-Engele P , Lim LP and Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007; 27: 91 -105. [PubMed] .
- 32. Krichevsky AM , Sonntag KC , Isacson O and Kosik KS. Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells. 2006; 24: 857 -864. [PubMed] .
- 33. Okita K , Ichisaka T and Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007; 448: 313 -317. [PubMed] .
- 34. Prozorovski T , Schulze-Topphoff U , Glumm R , Baumgart J , Schroter F , Ninnemann O , Siegert E , Bendix I , Brustle O , Nitsch R , Zipp F and Aktas O. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat Cell Biol. 2008; 10: 385 -394. [PubMed] .
- 35. Naguibneva I , Ameyar-Zazoua M , Polesskaya A , Ait-Si-Ali S , Groisman R , Souidi M , Cuvellier S and Harel-Bellan A. The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat Cell Biol. 2006; 8: 278 -284. [PubMed] .
- 36. Fulco M , Schiltz RL , Iezzi S , King MT , Zhao P , Kashiwaya Y , Hoffman E , Veech RL and Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003; 12: 51 -62. [PubMed] .
- 37. Li QJ , Chau J , Ebert PJ , Sylvester G , Min H , Liu G , Braich R , Manoharan M , Soutschek J , Skare P , Klein LO , Davis MM and Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007; 129: 147 -161. [PubMed] .
- 38. Kwon HS , Brent MM , Getachew R , Jayakumar P , Chen LF , Schnolzer M , McBurney MW , Marmorstein R , Greene WC and Ott M. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe. 2008; 3: 158 -167. [PubMed] .
- 39. Bhattacharyya SN , Habermacher R , Martine U , Closs EI and Filipowicz W. Stress-induced reversal of microRNA repression and mRNA P-body localization in human cells. Cold Spring Harb Symp Quant Biol. 2006; 71: 513 -521. [PubMed] .
- 40. Masuda K , Marasa B , Martindale JL , Halushka MK and Gorospe M. Tissue- and age-dependent expression of RNA-binding proteins that influence mRNA turnover and translation. Aging. 2009; 1: 681 -698. [PubMed] .
- 41. Marasa BS , Srikantan S , Martindale JL , Kim MM , Lee EK , Gorospe M and Abdelmohsen K. MicroRNA profiling in human diploid fibroblasts uncovers miR-519 role in replicative senescence. Aging. 2010; 2: .
- 42. Westerheide SD , Anckar J , Stevens SM Jr , Sistonen L and Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009; 323: 1063 -1066. [PubMed] .
- 43. Christians E , Davis AA , Thomas SD and Benjamin IJ. Maternal effect of Hsf1 on reproductive success. Nature. 2000; 407: 693 -694. [PubMed] .
- 44. Wang RH , Sengupta K , Li C , Kim HS , Cao L , Xiao C , Kim S , Xu X , Zheng Y , Chilton B , Jia R , Zheng ZM , Appella E , Wang XW , Ried T and Deng CX. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008; 14: 312 -323. [PubMed] .
- 45. Jin XL , Chandrakanthan V , Morgan HD and O'Neill C. Preimplantation embryo development in the mouse requires the latency of TRP53 expression, which is induced by a ligand-activated PI3 kinase/AKT/MDM2-mediated signaling pathway. Biol Reprod. 2009; 81: 234 -242. [PubMed] .
- 46. Chua KF , Mostoslavsky R , Lombard DB , Pang WW , Saito S , Franco S , Kaushal D , Cheng HL , Fischer MR , Stokes N , Murphy MM , Appella E and Alt FW. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005; 2: 67 -76. [PubMed] .
- 47. Fujita Y , Kojima K , Hamada N , Ohhashi R , Akao Y , Nozawa Y , Deguchi T and Ito M. Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem Biophys Res Commun. 2008; .
- 48. Yamakuchi M , Ferlito M and Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008; 105: 13421 -13426. [PubMed] .
- 49. Shi L , Cheng Z , Zhang J , Li R , Zhao P , Fu Z and You Y. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008; 1236: 185 -193. [PubMed] .
- 50. Pekarsky Y , Santanam U , Cimmino A , Palamarchuk A , Efanov A , Maximov V , Volinia S , Alder H , Liu CG , Rassenti L , Calin GA , Hagan JP , Kipps T and Croce CM. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006; 66: 11590 -11593. [PubMed] .
- 51. Cummins JM , He Y , Leary RJ , Pagliarini R , Diaz LA Jr , Sjoblom T , Barad O , Bentwich Z , Szafranska AE , Labourier E , Raymond CK , Roberts BS , Juhl H , Kinzler KW , Vogelstein B and Velculescu VE. The colorectal microRNAome. Proc Natl Acad Sci U S A. 2006; 103: 3687 -3692. [PubMed] .
- 52. Fabbri M , Garzon R , Cimmino A , Liu Z , Zanesi N , Callegari E , Liu S , Alder H , Costinean S , Fernandez-Cymering C , Volinia S , Guler G , Morrison CD , Chan KK , Marcucci G , Calin GA , Huebner K and Croce CM. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007; 104: 15805 -15810. [PubMed] .
- 53. Yanaihara N , Caplen N , Bowman E , Seike M , Kumamoto K , Yi M , Stephens RM , Okamoto A , Yokota J , Tanaka T , Calin GA , Liu CG , Croce CM and Harris CC. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006; 9: 189 -198. [PubMed] .
- 54. Lavu S , Boss O , Elliott PJ and Lambert PD. Sirtuins--novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008; 7: 841 -853. [PubMed] .
- 55. Nichols J , Evans EP and Smith AG. Establishment of germ-line-competent embryonic stem (ES) cells using differentiation inhibiting activity. Development. 1990; 110: 1341 -1348. [PubMed] .
- 56. Bain G , Kitchens D , Yao M , Huettner JE and Gottlieb DI. Embryonic stem cells express neuronal properties in vitro. Dev Biol. 1995; 168: 342 -357. [PubMed] .
- 57. Zhao Y , Samal E and Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005; 436: 214 -220. [PubMed] .
- 58. Emiliani S , Fischle W , Van Lint C , Al-Abed Y and Verdin E. Characterization of a human RPD3 ortholog, HDAC3. Proc Natl Acad Sci U S A. 1998; 95: 2795 -2800. [PubMed] .
- 59. Fischle W , Dequiedt F , Fillion M , Hendzel MJ , Voelter W and Verdin E. Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J Biol Chem. 2001; 276: 35826 -35835. [PubMed] .
- 60. Gao GP , Alvira MR , Wang L , Calcedo R , Johnston J and Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002; 99: 11854 -11859. [PubMed] .