Introduction
Disruption in metabolic homeostasis and
over accumulation of metabolites, cholesterol, bile acids, triglycerides (fat),
or glucose, play causative roles in the development of metabolic disorders,
such as, atherosclerosis and related heart disease, fatty liver, obesity, and
diabetes. The NAD+-dependent SIRT1 deacetylase plays a critical role in
maintaining metabolic homeostasis which affects aging so that SIRT1 increases
life spans in most organisms, including mammals [1-3]. Despite extensive
studies on SIRT1 function and its beneficial metabolic effects, how the
expression of SIRT1 is regulated under normal conditions and how SIRT1 levels
are decreased in metabolic disease states remain unclear. In this review, we
survey recent studies showing how SIRT1 expression is regulated at the
post-transcriptional level, focusing on microRNAs (miRs) which have recently
emerged as important cellular regulators [4-6]. We also review recent studies
showing that the nuclear receptor FXR/SHP cascade pathway which controls
expression of miR-34a and its target SIRT1 in normal conditions and is
dysregulated in metabolic disease states.
SIRT1: a key regulator in cellular metabolism
Caloric restriction (CR) was shown to increase life
span and promote survival in yeast, worms, flies, rodents and perhaps primates
[1,2]. SIRT1 mediates the beneficial metabolic effects of CR in an NAD+-dependent
manner by deacetylating and altering the activities of transcriptional factors
which regulate metabolic genes [1,2,7]. SIRT1 deacetylates and activates
transcript-tional ability of metabolic regulators, such as PGC-1α, p53,
Foxo 1, NF-κB, LXR, and FXR that are involved in lipid and glucose
metabolism, inflammation, mitochondrial biogenesis, and energy balance [1,2,8-12]. In addition, SIRT1 was shown to be recruited to the promoter of metabolic
target genes and suppress their transcription [13,14]. It was reported that
SIRT1 is associated with the promoter of PPARγ, a key adipogenic factor,
and suppresses PPARγ transcription by recruiting the corepressors, NcoR1
and SMRT [14]. SIRT1 was reported to bind to the UCP 2 gene promoter and
inhibit its transcription in pancreatic β-cells, resulting in increased
ATP production and insulin secretion [13]. SIRT1 was also shown to improve
insulin sensitivity by repressing transcription of protein tyrosine phosphatase
1B, a major negative regulator of insulin action, via histone deacetylation
[15]. Beneficial metabolic functions of SIRT1 have been demonstrated in studies
using small molecule activators and transgenic mice that are null for SIRT1 or
overexpress SIRT1 [16-20]. The natural compound resveratrol and the synthetic
compound SRT1720 are activators of SIRT1 and have been shown to ameliorate
insulin resistance, increase mitochondrial content, improve metabolic profiles,
and increase survival in mice fed a high-fat diet [16-18]. Transgenic mice
expressing SIRT1 were shown to be resistant to body weight gain and ameliorated
insulin resistance and glucose intolerance in these mice compared to wild-type
control mice [20]. Further, transgenic mice expressing moderate amounts of
SIRT1 were also shown to protect livers from diet-induced metabolic damage [12,21]. Consistent with these reports, in liver-specific SIRT1 null mice
challenged with a high fat diet, fatty acid metabolism was altered and the
development of fatty livers and inflammatory responses were promoted [19,22].
Loss of function studies also showed that SIRT1 decreases endothelial
activation in hypercholesterolemic ApoE-/- mice without affecting
endothelium-dependent vasodilatation [23]. All these recent studies demonstrate
that SIRT1 is a key regulator of cellular metabolism and mediates beneficial
metabolic effects.
MicroRNAs: emerging metabolic regulators
MicroRNAs (miRNAs) are small
(approximately 22 nt) non-coding RNAs that control gene expression [4-6]. MiRs
are transcribed from DNA by RNA polymerase II as hairpin precursors which are
further processed to mature forms [4-6]. MiRs bind to the 3'-untranslated
region (UTR) of target mRNAs and inhibit their expression by causing mRNA
cleavage or inhibition of translation. Approximately 30% of all human genes are
thought to be regulated by miRs [5,6] and indeed, miRs control gene expression
in diverse biological processes including development, differentiation, cell
prolifera-tion, and apoptosis. Recent studies have demonstrated crucial roles of
miRNAs in the regulation of cellular metabolism [24-32]. MiRs are involved in
lipid and glucose metabolism in major metabolic tissues, such as, liver,
pancreas, adipose, and muscle as summarized in Table 1. Mir-122 is the most
abundant miR in the liver and plays important roles in a wide variety of liver
functions ranging from cholesterol metabolism, liver cancer, stress responses,
viral infection, to circadian regulation of hepatic genes [24,28,29]. MiR-33
has been shown to contribute to the regulation of cholesterol homeostasis by
targeting the cholesterol transporter genes, ABCA1 and ABCG1 [25,26]. Our
group recently reported that miR-34a targets hepatic SIRT1 and, interestingly,
expression of miR-34a was highly elevated and SIRT1 levels were decreased in
fatty livers of diet-induced obese mice [30]. MiR-34a was also shown to
suppress insulin secretion in pancreatic β-cells [33]. The roles of
miR-375 in pancreatic islet functions, especially in insulin gene
transcription, insulin secretion, and islet cell growth, are also well
established [31,32]. Mir-27 and miR-378 were reported to control adipocyte
differentiation and lipid synthesis, respectively [34,35]. MiR-223 was shown
to regulate glucose uptake in cardiomyocytes and miR-696 to regulate
mitochondria biogenesis and fatty acid oxidation in gastrocnemius muscle [36,37]. In line with their critical functions, miRs are often underexpressed or
overexpressed in disease states [4,6,24,28,30,38-40]. Recent studies have shown
that restoring miRs or downregulating miRs using antisense miR inhibitors,
called antagomirs, has improved transcriptional and biological outcomes,
demonstrating that miRs are promising therapeutic targets [4,24,38].
Down-regulation of SIRT1 by microRNAs
Consistent with its critical roles in diverse
biological processes, the regulation of SIRT1 expression is fine tuned at
multiple levels, including transcriptional, post-transcriptional, and post-translational
levels. The general regulation of SIRT1 activity and expression has been thoroughly
reviewed in excellent articles [1-3,41] and, therefore, this review focuses on
the regulation of SIRT1 expression by miRs (Table 2). MiR-34a was first
identified as a posttranscriptional regulator of SIRT1 in the regulation of
apoptosis under cellular genotoxic stress in human colon cancer HCT116 cells
[42]. MiR-34a binds to the 3' UTR of SIRT1 mRNA in a partial complementary
manner and represses its translation but does not affect mRNA degradation [30,42]. Our group further reported that miR-34a targets hepatic SIRT1 in the regulation
of cellular metabolism in human hepatoma HpeG2 cells and in mouse liver in vivo
using adenoviral-mediated overexpression of miR-34a [30]. Remarkably, we
observed that miR-34a levels are highly elevated and SIRT1 protein levels are
substantially decreased in the fatty livers of both diet-induced obese mice and
the leptin-deficient ob/ob mice [30]. These findings are in line with recent
studies showing that miR-34a is the most elevated miR in livers exhibiting nonalcoholic
steatohepatitis, a spectrum of nonalcoholic fatty liver diseases in humans
[39]. Other miRs also target SIRT1. In response to nutritional availability,
miR-132 was shown to downregulate SIRT1, resulting in activation of
inflammatory pathways in adipose tissues [43]. MiR-199a was identified as a
negative regulator of SIRT1 and HIF1a, a key mediator of hypoxia [44]. Low
oxygen tension results in acute
downregulation of miR-199a in cardiac myocytes and in porcine heart and this
reduction is required for upregulation of its targets, HIF-1a and SIRT1 in
response to decreased oxygen [44]. Interestingly, a recent study showed that
SIRT1 protein levels are much higher in mouse embryonic stem cells (ESCs) than in
differentiated tissues and that miRNAs, miR-181a and b, miR-9, miR-204,
miR-199b, and miR-135, post-transcriptionally down-regulate SIRT1 during mouse
ESC differentiation and maintain low levels of SIRT1 expression in
differentiated tissues [45].
Table 1. MicroRNAs regulating cellular metabolism in major metabolic tissues.
| MicroRNA | Direct targets[putative] | Functions in Metabolism (references) | Tissues (cultured cells) |
| miR-33 |
ABCA1,
NPC1
|
Cholesterol
homeostasis (25, 26)
|
Liver
(HepG2)
|
| miR-34a |
SIRT1
|
lipid
metabolism, promotes fatty liver (30)
|
| miR-370 |
Cpt1a
|
Fatty
acid and triglyceride biosynthesis (29)
|
| miR-122 |
CAT-1
ADAM17
|
Hepatic
lipid metabolism (24, 29)
Circadian
gene expression (28)
|
| miR-34a |
VAMP2
|
B-cell
exocytosis (33)
|
Pancreatic
Islets
(MIN6,
INS-1)
|
| miR-124a |
Foxa2
|
Intracellular
signaling in pancreatic β-cell (27)
|
| miR-375 |
MTPN
|
Regulates
catecholamine release
Inhibits
insulin secretion (31, 32)
|
| miR-27a |
[PPARγ,
C/EBPα]
|
Inhibits
adipocyte formation,
Down-regulated
during adipogenic differentiation (34)
|
(Adipocytes,
3T3-L1, ST2)
|
| miR-378/378* |
[Ribosomal
proteins]
|
Upregulates
adipocyte differentiation and lipid synthesis (35)
|
| miR-223 |
Glut4
|
Glucose
uptake and insulin resistance (36)
|
Muscle Gastrocnemius
(Cardiomyocyte,
C2C12)
|
| miR-696 |
[PGC1α]
|
Muscle
metabolism, mitochondria biogenesis and fatty acid oxidation (37)
|
Table 2. MicroRNAs targeting SIRT12.
| MicroRNA | Sequences of microRNAs | Size (nt) | Biological functions (references) |
| miR-34a |
5'-uggcagugucuuagcugguugu-3'
|
22
|
Hepatic
lipid metabolism (30)
Islet
β-cell exocytosis (33)
Cell apoptosis (42)
|
| miR-132 |
5'-uaacagucuacagccauggucg-3'
|
22
|
Stress-induced
chemokine production (43)
|
| miR-199a |
5'-cccaguguucagacuaccuguuc-3'
|
25
|
Hypoxia preconditioning
(44)
|
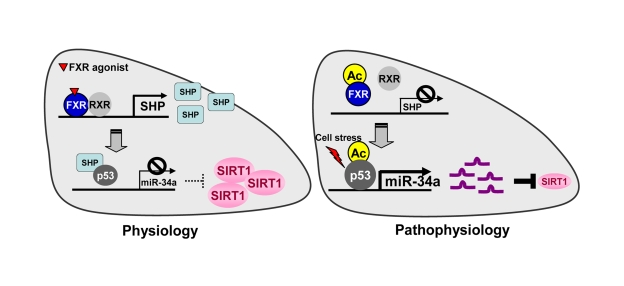
Figure 1. The FXR/SHP pathway controlling miR-34a and SIRT1 expression. Under normal
conditions, activation of FXR signaling induces the metabolic repressor SHP
in liver. SHP is then recruited to the miR-34a promoter and inhibits
binding of the key activator p53 to the DNA, resulting in decreased miR-34a
expression. Inhibition of miR-34a results in increased hepatic SIRT1 levels.
In contrast, under pathophysiological conditions such as fatty livers of
obese mice, the dysregulated FXR/SHP pathway due to highly elevated FXR
acetylation no longer inhibits transcription of miR-34a. The dysregulated
FXR/SHP pathway, along with acetylation of p53 due to cellular stress under
metabolic disease states, result in elevated miR-34a expression, which
contributes to decreased SIRT1 levels.
A novel FXR/SHP/miR-34a pathway controlling SIRT1
levels
The nuclear bile acid receptor, Farnesoid
X Receptor (FXR), plays an important role in maintaining lipid and glucose
levels by regulating expression of numerous metabolic genes mainly in the liver
and intestine [46]. Consistent with its important metabolic functions, disruption
of the FXR gene in transgenic mice was associated with metabolic diseases,
including hypercholesterolemia, cholesterol gallstone disease, fatty liver, and
type 2 diabetes [46-49]. Activation of FXR in diabetic obese mice improved
metabolic outcomes by reducing serum glucose and lipid levels [50]. Although
both FXR and SIRT1 have been shown to be critical for hepatic metabolism and activation
of both proteins improves metabolic outcomes in diet-induced obese mice [17,18,46,47,50], it was unknown whether the expression and activity of these
two proteins are coordinately regulated. In recent studies, we found that FXR
positively regulates hepatic SIRT1 expression by inhibiting expression of
miR-34a [30]. As shown in Figure 1, under normal conditions, miR-34a levels are
down-regulated by a nuclear receptor cascade pathway involving FXR and orphan
nuclear receptor and metabolic repressor, Small Heterodimer Partner (SHP) [51,52]. Upon induction by activated FXR, SHP is recruited to the miR-34a promo- ter and suppresses its transcription by inhibiting the
promoter occupancy of p53, the key activator of the miR-34a gene [53].
Subsequently, inhibition of miR-34a contributes to increased expression of SIRT1.
This FXR/SHP pathway was also shown to play a crucial role in the regulation of
hepatic bile acid synthesis by inhibiting the rate-limiting bile acid synthetic
enzyme CYP7A1 [51,52] and to suppress fatty liver formation by inhibiting the
key lipogenic activator SREBP-1c [54]. Our group has identified molecular
mechanisms by which SHP inhibits its target genes by coordinately recruiting
chromatin modifying repressive cofactors, including HDACs, G9a metyltransferase,
and Brm-containing Swi/Snf remodeling complex [55-57]. Consistent with these
previous findings, we observed recruitment of HDACs to the miR-34a promoter in
mouse liver after treatment with the synthetic FXR agonist, GW4064 (not shown).
In contrast, in fatty livers of obese mice, the FXR/SHP pathway is dysregulated
such that miR-34a levels are highly elevated, which contributes to reduced
SIRT1 levels [30]. Interestingly, activation of FXR signaling in obese mice by
daily treatment with GW4064 for 5 days or by hepatic expression of FXR using
adenoviral delivery decreased miR-34a levels and restored SIRT1 levels [30]. Consistent
with a critical role for FXR in positively controlling SIRT1 through the inhibition
of miR-34a, miR-34a levels were indeed elevated and SIRT1 protein levels are
substantially decreased in FXR null mice [30]. Our findings suggest an
intriguing link among FXR activation, decreased miR-34a levels, increased SIRT1
levels, and beneficial metabolic outcomes.
A positively interacting FXR/SIRT1 regulatory loop
In the FXR/SHP/miR-34a pathway, FXR positively
regulates hepatic SIRT1 levels by inhibiting transcription of the miR-34a gene.
These findings, along with previous studies showing the p53/miR-34a/SIRT1
feedback loop [42,58], suggest intriguing regulatory loops controlling SIRT1
expression (Figure 2). In the short regulatory loop, SIRT1 positively
auto-regulates its own expression by deacetylating p53 and histones at the
miR-34a promoter, resulting in suppression of miR-34a [9,30,42,53,58]. In
the long regulatory loop, SIRT1-mediated deacetylation of FXR increases FXR's
transactivation ability by increasing binding of the FXR/RXR heterodimer to DNA
resulting in induction of SHP and repression of miR-34a expression [11,30]. We
observed that FXR acetylation is dynamically controlled by p300 acetylase and
SIRT1 deacetylase under normal conditions, and remarkably,
FXR acetylation levels are highly elevated in fatty livers of obese mice [11].
Interestingly, treatment daily with the SIRT1 activator resveratrol for 1 week
or adenoviral-mediated hepatic expression of SIRT1 substantially reduced FXR
acetylation with beneficial metabolic effects [11]. These results are
consistent with the idea that the transactivation activity of FXR is low in
obese mice due to highly elevated FXR acetylation, which contributes to
increased expression of miR-34a. Subsequently, elevated miR- 34a suppresses
expression of SIRT1, which then further decreases FXR activity, resulting in a vicious
FXR/miR-34a/SIRT1 regulatory loop in metabolic disease states. In addition to deacetylation
of FXR, SIRT1 has been implicated as a positive regulator of the expression and
activity of FXR. During fasting, PGC-1α was shown to increase expression
of the FXR gene and function as a coactivator of FXR [59]. Since SIRT1
deacetylates and increases PGC-1α activity [8], SIRT1 should increase FXR
expression and activity by enhancing PGC-1α activity. All these recent
studies strongly suggest that the expression and activity of these two proteins
are mutually and coordinately regulated.
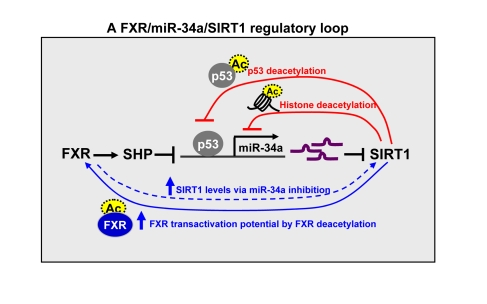
Figure 2. A FXR/SIRT1
positive-feedback regulatory loop. The expression and activity of FXR
and SIRT1 are mutually and coordinately regulated. SIRT1 positively
auto-regulates its own expression by inhibiting miR-34a via deacetylation
(as indicated by dotted circles) of p53 and histones at the miR-34a
promoter (short loop) and by increasing transactivation potential of FXR
via deacetylating the FXR (long loop). SIRT1 also increases FXR expression
and activity via deacetylation of PGC-1α.
FXR in turn positively regulates hepatic SIRT1 expression by inhibiting
miR-34a which targets SIRT1.
Concluding remarks
Because of SIRT1's anti-aging properties and its
beneficial effects on a wide range of age-related disease [1-3,21], it has
been intensively studied. SIRT1 levels were reported to be decreased in liver,
muscle, and adipose tissues of diet-induced obese mice in vivo as well as in
cultured cell models of insulin resistance [15,30,60], but the underlying
mechanisms remain unclear. The discovery of the FXR/miR-34a pathway controlling
SIRT1 levels provides a partial explanation since elevated miR-34a levels in
obese mice contribute to decreased SIRT1 levels [30]. Based on these findings,
together with the development of effective inhibitors of miRs, the antagomirs [4,24,38], it will be interesting to see whether the reduction of elevated
miR-34a in fatty livers of obesity improves transcriptional profiles of
metabolic genes and metabolic outcomes. Also, it will be important to understand how the FXR/SIRT1 regulatory network is
dysregulated in metabolic disease states which likely involves altered cellular
kinase signaling pathways that post-transcriptionally affect SIRT1 and FXR
levels and activities. Development of drugs that target the FXR/miR-34a pathway
and other miRs controlling SIRT1 expression may lead to novel therapeutic
options for treating age-related metabolic disease including fatty liver,
obesity and type II diabetes.