



Introduction
Diabetes mellitus is the most prevalent metabolic disorder, and its metabolic hallmark, glucose intolerance, affects approximately 344 million people worldwide [1]. There are two prominent subtypes of diabetes mellitus, named type 1 and type 2, respectively. Both sub-types affect the production and secretion of insulin from pancreatic beta-cells. While type 1 diabetes is considered an autoimmune disease leading to slowly progressive and ultimately complete loss of pancreatic beta-cells, type 2 diabetes is caused by a combination of peripheral insulin resistance and beta-cell dysfunction. Recent evidence however suggests that type 2 diabetes is additionally characterized by impaired beta-cell regeneration and reduced beta-cell mass [2-5].
Eukaryotic chromosomes carry tandemly repeated terminal sequences, so-called telomeres, which are essential for chromosome stability. Telomeric DNA is synthesized, elongated and hence maintained by copying an RNA template sequence within the RNA moiety of an enzyme called telomerase. The respective subunit of telomerase is encoded by the telomerase RNA component (Terc) gene. Significant evidence suggests that telomeres and telomerase activity have a crucial role in regulating cell survival and regeneration [6-8].
Given the role of telomerase in maintenance and replication of differentiated cells on the one hand, and the importance of beta-cell loss for the pathogenesis of type 1 as well as type 2 diabetes on the other hand, it appears tempting to hypothesize that telomerase could act as key regulator of beta-cell viability and regeneration. We therefore here have analyzed previously generated mice [9,10] that are deficient for the telomerase RNA component (Terc) gene in this regard. We find that these animals develop glucose intolerance and impaired insulin secretion due to impaired beta-cell regeneration.
Results
Body composition and energy expenditure in Terc-/- G4 mice
As previously reported [9,10], Terc -/- G4 mice are smaller and have reduced body mass (Figure 1A). However, we here show that this reduction affects both lean mass (Figure 1B) and body fat (Figure 1C) to the same extent. We moreover demonstrate that both dietary energy uptake (Figure 1D) and energy excretion via faeces (Figure 1E) is reduced. However when these latter findings are normalized to body mass, no significant differences can be found for neither energy uptake nor energy excretion (data not shown). Furthermore, no glucosuria was detected in none of the genotypes (data not shown), precluding differential loss of energy via urine. Accordingly determination of energy expenditure as quantified by indirect calorimetry did not show any differences regarding energy turnover at neither day- nor night-time (Figure 1F).
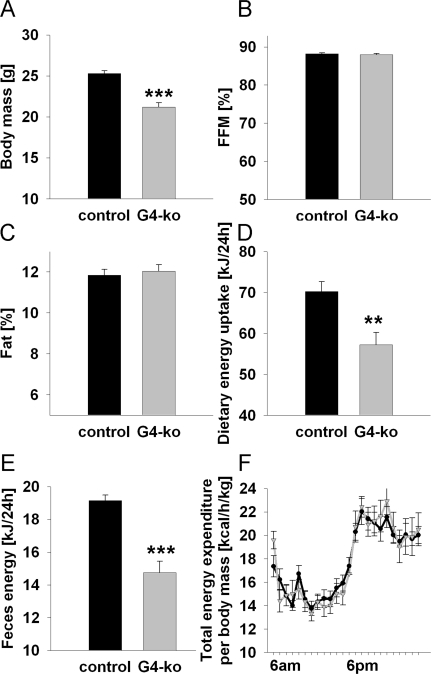
Figure 1. Impaired telomerase activity - Effects on body mass, body composition and energy expenditure. (A) Depicts body mass of 19-24 week old Terc-/- G4 animals (grey) and corresponding controls (black) (n=60 controls, n=61 Terc-/- G4). (B) Relative fat-free mass and (C) relative body fat content in mice as in Panel A (n=60 controls, n=61 Terc-/- G4). (D) Food uptake per individual mouse, depicted for mice as in Panel A (n=30 controls, n=10 Terc-/- G4). (E) Energy excreted via faeces as quantified by direct bomb calorimetry (n=30 controls, n=10 Terc-/- G4). (F) Total energy expenditure normalized to body mass of mice as in Panel A (n=10 controls, n=10 Terc-/- G4).
Taken together these findings indicate that Terc -/- G4 mice are smaller but have an unaltered relative body composition and energy metabolism.
Metabolic plasma markers in Terc -/- G4 mice
We quantified concentrations of glucose (fasted and fed states), insulin (fasted and fed states), triglycerides, non-esterified fatty acids, total cholesterol and alanine aminotransferase (all fasted) in plasma samples of the animals, and found no differences between Terc -/- G4 and control genotypes (Suppl. Figure 1).
Glucose metabolism in Terc -/- G4 mice
We next performed intra-peritoneal glucose tolerance tests (ipGTT) on Terc -/- G4 and on the control genotypes, and observed impaired glucose clearing in Terc -/- G4 mice (Figure 2A). We then aimed to elucidate whether impaired insulin secretion or rather insulin resistance may be responsible for this phenotype. We therefore first quantified plasma insulin levels during conditions of ipGTT, and observed impaired insulin secretion in response to injected glucose in Terc -/- G4 mice (Figure 2B). By contrast, performing intra-peritoneal insulin tolerance tests (ipITT) by injecting insulin into mice did not reveal significant differences in glucose disposal in response to insulin (Figure 2C). Since telomerase has been reported to affect mitochondrial function [11,12], we lastly quantified glucose oxidation rates by injecting uniformly 13C-labeled glucose to quantify the exhalation of 13CO2, which was found to be identical in all genotypes (Figure 2D), precluding systemic mitochondrial dysfunction in Terc -/- G4 mice, at least in regards to glucose-derived pyruvate oxidation capacity.
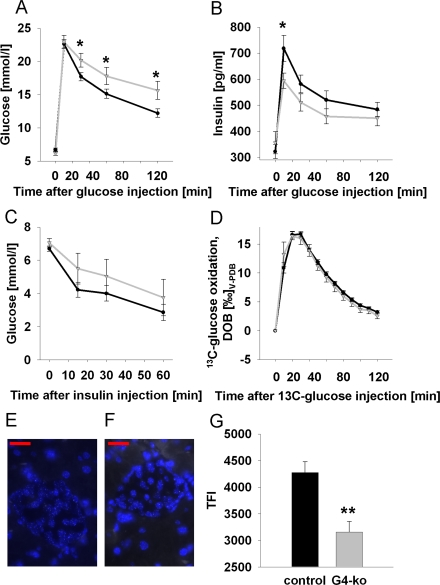
Figure 2. Impaired telomerase activity impairs glucose tolerance and glucose-stimulated insulin secretion. (A) Serum glucose excursions following intraperitoneal injection of D-glucose; black line corresponds to control animals while grey line reflects Terc-/- G4 animals (also applies to panels B-D) (n=47 controls, n=25 Terc-/- G4). (B) Plasma insulin excursions following intraperitoneal injection of D-glucose (n=47 controls, n=25 Terc-/- G4). (C) Blood glucose excursion following intraperitoneal injection of insulin (n=24 controls, n=8 Terc-/- G4). (D) Exhalation of 13Carbon dioxide per constant time interval following intraperitoneal injection of 13C-labeled glucose (n=23 controls, n=8 Terc-/- G4). Typical quantitative fluorescence in situ hybridization (qFISH) appearances of a control (E) and Terc -/- G4 (F) islet, respectively. (G) depicts results of telomere length quantification by qFISH (n=23 control and n=10 Terc-/- G4 animals, 25 nuclei per mouse were evaluated; red bars are 20 μm of length).
Since the phenotype appeared to be restricted to impaired insulin secretion, we next quantified telomere length specifically in pancreatic islets. Using quantitative fluorescence in situ hybridization (qFISH) (Figure 2 E, F) we observe a pronounced reduction of average telomere length in islets of Terc -/- G4 animals (Figure 2G).
Taken together, telomerase deficiency causes shortened telomere length in pancreatic islets, and impaired glucose tolerance as well as impaired insulin secretion in mice.
Reduced beta-cell mass in Terc -/- G4 mice
Reduced insulin secretion is typically caused by reduced mass of the islets of Langerhans, which mainly contain beta cells. To decide whether this possibility may apply in regards to the phenotype observed (Figure 2 A, B), we first determined beta-cell mass in repeated sections from pancreata of all four genotypes by quantitative microscopy. We firstly observed smaller islets in mice deficient for Terc (Figure 3B) as compared to control animals (Figure 3A). By morphometrically quantifying insulin-positive areas within pancreatic sections of all genotypes, we consistently found a reduction of insulin-positive areas per total pancreatic section (Figure 3C). Since Terc -/- G4 are smaller than control genotypes (Figure 1A), we normalized pancreatic and insulin-positive areas to body mass to exclude the possibility that reduced insulin-positive area size simply was an indicator of reduced body mass. After normalizing the areas for body mass, we still observe a significant reduction of insulin-positive areas (Figure 3D).
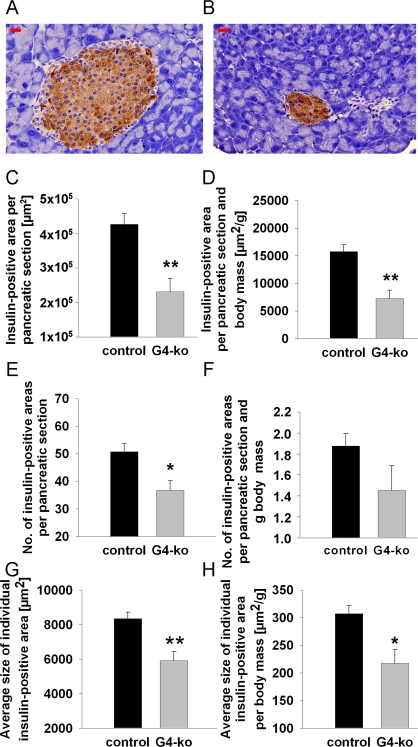
Figure 3. Impaired telomerase activity reduces number and size of insulin-containing pancreatic islets. (A) Depicts a typical control islet, while (B) reflects a typical islet from an Terc-/- G4 animal; red bars are 20 μm of length. (C, D) depict insulin-positive area size normalized to whole pancreatic section area only (C) and additionally normalized to body mass (D). (E, F) shows the number of islets per pancreatic section (E) and after additional normalization to body mass (F). (G, H) depict the sizes of individual insulin-positive areas per se (G) and after normalization to body mass (H). Black bars reflect control genotypes (n=25) and grey bars indicate Terc-/- G4 animals (n=10).
We moreover find the number of islets to be reduced in Terc -/- G4 animals (Figure 3E) which however did not apply after normalization for body mass (Figure 3F, P=n.s.). Most importantly, we find the insulin-positive area per individual islet of Langerhans to be reduced in absolute terms (Figure 3G) as well as after normalization for body mass (Figure 3H), altogether suggesting that telomerase deficiency causes a reduction of beta cell mass by reducing the size of individual islets whereas islet number is not affected.
Impaired beta-cell regeneration in Terc -/- G4 mice
We next injected bromo-desoxyuridine (BrdU) into mice prior to sacrifice which enables us to determine the number of DNA-replicating, i.e. dividing beta cells, after counterstaining with an anti-BrdU antibody. We found a reduction of BrdU-positive beta-cells per islet (Figure 4A) as well as after normalization for body mass (Figure 4B). Lastly, the number of BrdU-positive beta-cells per standardized insulin-positive area was also found to be reduced (Figure 4C) as well as after normalization for body mass (Figure 4D).
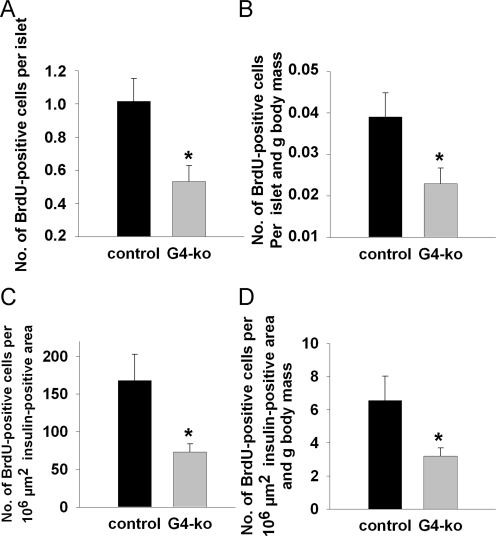
Figure 4. Impaired telomerase activity reduces islet number and size by impairing the replicative potential of pancreatic beta-cells. (A, B) shows the number of BrdU-positive cells per insulin-positive area (A) and after additional normalization to body mass (B). (C, D) depict the number of BrdU-positive cells per standardized individual insulin-positive area (C) and after additional normalization to body mass (D). Black bars reflect control genotypes (n=9) and grey bars indicate Terc-/- G4 animals (n=10).
Taken together, these findings indicate that telomerase deficiency as found in Terc -/- G4 animals causes reduced beta-cell mass due to impaired regenerative capacity of pancreatic islets of Langerhans, culminating in impaired glucose tolerance due to reduced insulin secretion in vivo.
Discussion
We here describe a direct causal link between impaired telomerase activity and impaired insulin secretion as well as glucose intolerance in Terc-/- G4 mice. These findings are supported by the fact that shortened telomeres, as found in states of reduced telomerase activity, are associated with type 2 [13-20] as well as possibly type 1 diabetes mellitus [21,22] in humans. As stated in the introductory section, both disorders exhibit patterns of impaired insulin secretion.
According to our findings, glucose intolerance in Terc-/- G4 animals is caused by reduced insulin secretion only, whereas insulin sensitivity was found to be unaffected. This latter observation is in conflict with previous reports that shortened telomeres in humans are associated with insulin resistance [14,23-25], a state in which insulin sensitivity is reduced. However and since insulin secretion and insulin sensitivity appear to influence each other primary disturbances in insulin secretion may secondarily affect insulin resistance.
Progressive beta-cell failure and senescence are hallmarks of type 2 diabetes [2-5]. More specifically, telomere shortening has been shown to determine the risk of beta-cell growth arrest and cellular senescence in adult human islet cells [26]. The Tertgene is moderately expressed in islets from different mouse models, including C57BL/6 (the strain used in the present study), and expression levels interestingly appear to decrease with age [27]. Moreover, significant telomerase activity can be detected in islet extracts from C57BL/6 mice [27]. Interestingly, telomerase appears to counteract the senescence-promoting effects of hyperglycemia, at least in vitro [28]. Moreover, several of the known downstream targets of telomerase have been identified to affect islet cell growth and regeneration [29,30], whereas a link to telomerase deficiency had not been established. Further studies will have to elucidate whether our current findings and these putative downstream effectors converge in deterioration of islet mass and hence development of diabetes mellitus.
Taken together, our findings indicate that reduced telomerase activity may be considered a cause of impaired glucose tolerance and hence diabetes mellitus. In full accordance with the role of this protein in multiple other cell types, telomerase is required to maintain the replicative potential of pancreatic beta-cells. Consistently, lack of telomerase activity causes beta-cell loss and subsequent deterioration of glucose metabolism, reflecting the increasing incidence of diabetes mellitus with increasing age.
Materials and Methods
Generation and maintenance of mice
Four genotypes of the previously described Terc -/- animals were studied: While Terc -/- G4 mice have been intercrossed for three generations with Terc-deficient littermates, Terc-/- G1 have parents that were both heterozygous for the Terc disruption. The latter served as controls, in addition to wild-type and heterozygous Terc +/- animals, since significant telomere shortening is absent in such Terc -/- G1 animals [9,10]. Tail-biopsy genomic PCR for detection of the Terc-/- allele was performed as previously described [10]. Animals were housed in air-conditioned rooms (temperature, 20 ± 2 °C; relative moisture, 50 - 60 %) under a 12-hour-light/dark schedule (lights on at 06:00 hrs) and had free access to standard chow (Ssniff R/M-H, Ssniff Spezialdiäten GmbH, Soest, Germany) and water. Mice were studied at an age of 19 - 24 weeks, and were kept in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals, and all experiments were approved the corresponding institutional review boards.
Body composition analysis
Body mass and body composition (including fat content) were measured by use of quantitative nuclear magnetic resonance technique (Echo MRI-100 Body Composition Analyzer, Echo Medical Systems, Houston, TX) as previously described [31].
Food consumption quantification
Food intake was recorded weekly and faeces were collected 3 times per week. After freeze-drying energy content of diet and faeces samples was determined by bomb calorimetry (IKA C5003, IKA Werke GmbH, Staufen, Germany) as previously described [31].
Respiratory quotient and energy expenditure
Total energy expenditure and respiratory quotient were determined by indirect calorimetry of mice housed individually in metabolic cages, receiving food and water ad libitum as described using a TSE LabMaster Calorimetry Module (TSE, Bad Homburg, Germany) [31].
Insulin and glucose tolerance tests
Glucose tolerance tests were performed as described [31] by intraperitoneal (ip) glucose injection of 1 g per kg body mass of D-(+)-glucose (Merck, Darmstadt, Germany) after mice were fasted 16 hrs overnight. Insulin tolerance tests were performed by intraperitoneal (ip) injection of 1 U insulin per kg body mass as described [32].
Plasma and serum analyses
All blood samples were obtained, prepared, stored and measured exactly as previously described [31].
13CO2 breath tests for glucose oxidation experiments
13CO2 breath tests were applied as non-invasive methods to study the oxidation of injected or ingested 13C-labeled glucose exactly as previously described [31].
Quantification of pancreas and islet sizes
All procedures, including immuno-histochemistry for insulin, BrdU-injection and detection were performed as described [32] while digitizing and quantification for beta-cell-mass was performed with a MiraxMidi slide scanner and the AutMess program of AxioVision 4.6 software (both from Carl Zeiss MicroImaging, Jena, Germany).
Telomere length
in pancreatic islets was determined on sections as above using quantitative fluorescence in situ hybridization (qFISH) according to previously reported methods [33]. The relative telomere length was determined by the fluorescence intensity using the TFL software package [34].
Data analysis
Data are expressed as mean ± SEM. Number of animals analyzed is given in the respective figure legends. Equal distributions were tested by Kolmogorov-Smirnov test before applying T-tests. Unpaired Student's T-tests were used to compare Terc-/- G4, Terc -/- G1, Terc +/- and wild-type animals by employing SPSS 15.0 (SPSS, Chicago, IL, USA).
Supplementary Materials
Acknowledgments
The excellent technical assistance of Elisabeth Meyer, Susann Richter, Waltraud Scheiding, and Elke Thom is gratefully acknowledged. We thank Dr. Wolfgang Meyerhof for the use of his MiraxMidi slide scanner. This work is part of the research program of the Jena Centre for Systems Biology of Ageing - JenAge funded by the German Ministry for Education and Research (Bundesministerium für Bildung und Forschung - BMBF; support code: 0315581[A-D]).
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Unwin N, Gan D, Mbanya JC, Ramachandran A, Roglic G, Shaw J, Soltesz G, Whiting D, Zgibor J, Zhang P, Zimmet P. 2009; IDF Diabetes Atlas Unwin N, Whiting D, Gan D, Jacqmain O, Ghyoot G. Brussels .
- 2. Porte D Jr and Kahn SE. Beta-cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes. 2001; 50: Suppl 1 S160 -163. [PubMed] .
- 3. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004; 429: 41 -46. [PubMed] .
- 4. Georgia S and Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004; 114: 963 -968. [PubMed] .
- 5. Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death? Science. 2005; 307: 380 -384. [PubMed] .
- 6. Blackburn EH. Telomerases. Annu Rev Biochem. 1992; 61: 113 -129. [PubMed] .
- 7. Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005; 6: 611 -622. [PubMed] .
- 8. Blagosklonny MV, Campisi J, Sinclair DA, Bartke A, Blasco MA, Bonner WM, Bohr VA, Brosh RM Jr, Brunet A, DePinho RA, Donehower LA, Finch CE, Finkel T, et al. Impact papers on aging in 2009. Aging. 2010; 2: 111 -121. [PubMed] .
- 9. Lee HW, Blasco MA, Gottlieb GJ, Horner JW 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998; 392: 569 -574. [PubMed] .
- 10. Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999; 96: 701 -712. [PubMed] .
- 11. Saretzki G. Telomerase, mitochondria and oxidative stress. Exp Gerontol. 2009; 44: 485 -492. [PubMed] .
- 12. Sahin E and DePinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010; 464: 520 -528. [PubMed] .
- 13. Adaikalakoteswari A, Balasubramanyam M, Mohan V. Telomere shortening occurs in Asian Indian Type 2 diabetic patients. Diabet Med. 2005; 22: 1151 -1156. [PubMed] .
- 14. Sampson MJ, Winterbone MS, Hughes JC, Dozio N, Hughes DA. Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care. 2006; 29: 283 -289. [PubMed] .
- 15. Adaikalakoteswari A, Balasubramanyam M, Ravikumar R, Deepa R, Mohan V. Association of telomere shortening with impaired glucose tolerance and diabetic macroangiopathy. Atherosclerosis. 2007; 195: 83 -89. [PubMed] .
- 16. Tentolouris N, Nzietchueng R, Cattan V, Poitevin G, Lacolley P, Papazafiropoulou A, Perrea D, Katsilambros N, Benetos A. White blood cells telomere length is shorter in males with type 2 diabetes and microalbuminuria. Diabetes Care. 2007; 30: 2909 -2915. [PubMed] .
- 17. Uziel O, Singer JA, Danicek V, Sahar G, Berkov E, Luchansky M, Fraser A, Ram R, Lahav M. Telomere dynamics in arteries and mononuclear cells of diabetic patients: effect of diabetes and of glycemic control. Exp Gerontol. 2007; 42: 971 -978. [PubMed] .
- 18. Olivieri F, Lorenzi M, Antonicelli R, Testa R, Sirolla C, Cardelli M, Mariotti S, Marchegiani F, Marra M, Spazzafumo L, Bonfigli AR, Procopio A. Leukocyte telomere shortening in elderly Type2DM patients with previous myocardial infarction. Atherosclerosis. 2009; 206: 588 -593. [PubMed] .
- 19. Salpea KD, Talmud PJ, Cooper JA, Maubaret CG, Stephens JW, Abelak K, Humphries SE. Association of telomere length with type 2 diabetes, oxidative stress and UCP2 gene variation. Atherosclerosis. 2010; 209: 42 -50. [PubMed] .
- 20. Zee RY, Castonguay AJ, Barton NS, Germer S, Martin M. Mean leukocyte telomere length shortening and type 2 diabetes mellitus: a case-control study. Transl Res. 2010; 155: 166 -169. [PubMed] .
- 21. Jeanclos E, Krolewski A, Skurnick J, Kimura M, Aviv H, Warram JH, Aviv A. Shortened telomere length in white blood cells of patients with IDDM. Diabetes. 1998; 47: 482 -486. [PubMed] .
- 22. Astrup AS, Tarnow L, Jorsal A, Lajer M, Nzietchueng R, Benetos A, Rossing P, Parving HH. Telomere length predicts all-cause mortality in patients with type 1 diabetes. Diabetologia. 2010; 53: 45 -48. [PubMed] .
- 23. Aviv A, Valdes A, Gardner JP, Swaminathan R, Kimura M, Spector TD. Menopause modifies the association of leukocyte telomere length with insulin resistance and inflammation. J Clin Endocrinol Metab. 2006; 91: 635 -640. [PubMed] .
- 24. Demissie S, Levy D, Benjamin EJ, Cupples LA, Gardner JP, Herbert A, Kimura M, Larson MG, Meigs JB, Keaney JF, Aviv A. Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell. 2006; 5: 325 -330. [PubMed] .
- 25. Gardner JP, Li S, Srinivasan SR, Chen W, Kimura M, Lu X, Berenson GS, Aviv A. Rise in insulin resistance is associated with escalated telomere attrition. Circulation. 2005; 111: 2171 -2177. [PubMed] .
- 26. Halvorsen TL, Beattie GM, Lopez AD, Hayek A, Levine F. Accelerated telomere shortening and senescence in human pancreatic islet cells stimulated to divide in vitro. J Endocrinol. 2000; 166: 103 -109. [PubMed] .
- 27. Liew CW, Holman A, Kulkarni RN. The roles of telomeres and telomerase in beta-cell regeneration. Diabetes Obes Metab. 2009; 11: Suppl 4 21 -29. [PubMed] .
- 28. Blazer S, Khankin E, Segev Y, Ofir R, Yalon-Hacohen M, Kra-Oz Z, Gottfried Y, Larisch S, Skorecki KL. High glucose-induced replicative senescence: point of no return and effect of telomerase. Biochem Biophys Res Commun. 2002; 296: 93 -101. [PubMed] .
- 29. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006; 443: 453 -457. [PubMed] .
- 30. Nyblom HK, Bugliani M, Fung E, Boggi U, Zubarev R, Marchetti P, Bergsten P. Apoptotic, regenerative, and immune-related signaling in human islets from type 2 diabetes individuals. J Proteome Res. 2009; 8: 5650 -5656. [PubMed] .
- 31. Kuhlow D, Zarse K, Voigt A, Schulz TJ, Petzke KJ, Schomburg L, Ristow M. Opposing effects of dietary sugar and saturated fat on cardiovascular risk factors and glucose metabolism in mitochondrially impaired mice. Eur J Nutr. 2010; in press, DOI 10.1007/s00394-010-0100-4 http://www.springerlink.com/content/q38138074x38112061q/ .
- 32. Ristow M, Mulder H, Pomplun D, Schulz TJ, Müller-Schmehl K, Krause A, Fex M, Puccio H, Müller J, Isken F, Spranger J, Müller-Wieland D, Magnuson MA, et al. Frataxin-deficiency in pancreatic islets causes diabetes due to loss of beta-cell mass. J Clin Invest. 2003; 112: 527 -534. [PubMed] .
- 33. Lechel A, Holstege H, Begus Y, Schienke A, Kamino K, Lehmann U, Kubicka S, Schirmacher P, Jonkers J, Rudolph KL. Telomerase deletion limits progression of p53-mutant hepatocellular carcinoma with short telomeres in chronic liver disease. Gastroenterology. 2007; 132: 1465 -1475. [PubMed] .
- 34. Poon SS, Martens UM, Ward RK, Lansdorp PM. Telomere length measurements using digital fluorescence microscopy. Cytometry. 1999; 36: 267 -278. [PubMed] .