



For the last two decades, many genes and pathways have been shown to regulate aging across phyla. Among these, impaired mitochondrial respiration extends the life span of yeast, C. elegans, Drosophila, and mice [1-10]. Although several studies have proposed underlying mechanisms by which diminished mitochondrial respiration promotes longevity, including gene expression changes, global metabolic shift, and changes in energy utilization [11-15], the key genetic factors that mediate this long life span were poorly understood. We proposed that longevity caused by inhibition of mitochondrial respiration in C. elegans is promoted by HIF-1 activity via reactive oxygen species (ROS) [16]. Here, we discuss recent findings regarding the roles of HIF-1, ROS, and mitochondrial respiration in the regulation of aging.
The role of HIF-1 in aging
HIF-1 in mammalian cellular senescence
HIF-1α (hypoxia-inducible factor 1α) has been identified as the master regulator for cellular adaptation to hypoxia [17]. Under normal oxygen conditions, HIF-1α is maintained in a hydroxylated state by the HIF prolyl hydroxylase (prolyl hydroxylase-domain protein, PHD), leading to degradation of HIF-1, which is mediated by the E3 ubiquitin ligase von Hippel Lindau (VHL). Under low oxygen concentrations, the HIF prolyl hydroxylase does not hydroxylate HIF-1α. Therefore, HIF-1α is stabilized and translocated to the nucleus, where it forms a complex with HIF-1β. This HIF-1 complex binds to HIF-responsive elements (HREs) and turns on various genes to evoke immediate and long-term responses to hypoxia [17, 18]. This HIF-1-mediated transcriptional response has been shown to be crucial for many physiological processes, such as the adaptive response to hypoxia, angiogenesis, vasculogenesis, axon guidance, and aging [17-20].
Since it was first shown in the 1970s that exposing mammalian cells to low oxygen conditions extends their cellular life span [21], extensive research has been performed to understand the role of hypoxia and HIF-1 in cellular aging. In vivo microenvironment for hematopoietic stem cells is hypoxic, and stabilized HIF-1α is required to maintain their stem cell-like properties [22]. Mesenchymal stem cells cultured at an oxygen concentration of 3% showed delayed replicative senescence compared with cells cultured in ambient atmospheric conditions of ~20% O2 [23]. It has also been shown that aged cells display a decreased ability to express HIF-1 target genes under hypoxic conditions [24] and impaired binding of HIF-1 to HREs [25]. These observations may explain the susceptibility of aged organisms to hypoxic stress [24, 25]. Together these studies suggest that oxygen limitation and/or activation of HIF-1 play important roles in cellular senescence.
Regulation of the life span of C. elegans by HIF-1
Recent studies using C. elegans revealed the role of HIF-1 as a regulator of aging in the animal [16, 19, 26-32]. Initial characterization of hif-1 mutants showed that C. elegans also requires the HIF-1-dependent hypoxic response mechanism to adapt to hypoxic stress [33]. C. elegans hif-1 mutants are unique among multicellular model organisms because hif-1 null mutants are viable under normoxia [33]. Because of this viability, it has been straightforward to examine the role of HIF-1 in the regulation of life span.
When C. elegans is cultured in low oxygen, its life span is extended [34], raising the possibility that HIF-1 may promote the longevity of the animal. It has been possible to test this idea by asking whether up-regulation of HIF-1 can extend lifespan [16, 19, 26-32]. As in mammals, C. elegans EGL-9 (HIF prolyl hydroxylase) and VHL-1 (von Hippel Lindau 1, E3 ubiquitin ligase) are required for the degradation of HIF-1 [35,36]. Several groups have shown that down-regulation of vhl-1/E3 ubiquitin ligase or egl-9/HIF prolyl hydroxylase as well as overexpression of hif-1 significantly increases lifespan [16, 27, 29, 30, 32]. However, many issues regarding life span regulation by HIF-1 signaling remain unresolved. For example, the long life span of vhl-1/E3 ubiquitin ligase mutants was shown to be completely [30] or partially [32] dependent on hif-1 but in other reports the longevity caused by egl-9/HIF prolyl hydroxylase mutants was shown to be independent of hif-1[29]. Remarkably, several groups have reported that hif-1 deletion also extends lifespan [16, 26-29, 31]. Furthermore, the role of insulin/IGF-1 pathway [16, 26-28, 30] or dietary restriction [16, 28, 30] in the regulation of life span by HIF-1 signaling appear to be very complex [16, 19, 26-30, 32] (see Leiser and Kaeberlein 2010 for an extensive review on these complicated life span phenotypes caused by hif-1 signaling mutants [19]).
How can we resolve this controversy over the involvement of HIF-1 in signaling pathways modulating the life span of C. elegans? We propose that differences in the life span assay may largely account for this discrepancy. For example, one noticeable difference in experimental conditions is the temperature used for the life span measurement. Both Mehta et al. and our laboratory performed life span experiments at 20°C [16, 30], whereas Chen et al. introduced a temperature shift from 20°C to 25°C at the final larval stage prior to measuring adult life span [28]. Temperature is an environmental factor that critically influences life span: for example, worms live for a significantly shorter time at 25°C than at 20°C [37]. We previously showed that mutants with defects in thermosensory neurons have an even shorter life span at 25°C than wild-type animals and have a life span indistinguishable from that of wild type at 20°C [38]. From these data, we proposed that thermosensory neurons moderate the life span-shortening effect of the warm temperature (25°C) [38]. Interestingly, the temperature-dependent life span phenotype of hif-1 mutants is the opposite of that of the mutants with thermosensory defects: hif-1 mutants have a long life span when they are cultured at 25°C or shifted from 20°C to 25°C [16, 26, 28] and display no life span phenotypes at low temperatures (20°C and 15°C) [16, 26, 30]. Although this life span shortening is partly due to a temperature-dependent vulval integrity phenotype [26], the temperature-dependent longevity of hif-1 mutants remains largely intact even after excluding the contribution of the vulval integrity phenotype. Interestingly, HIF-1 has been shown to play a role in heat acclimation in C. elegans [39]. Therefore, it will be exciting to test whether this heat acclimation and/or thermosensation is involved in the effects of HIF-1 on the longevity response to temperature.
Mechanisms of the extension of life span by impaired mitochondrial respiration
The role of reactive oxygen species
Mild inhibition of the mitochondrial electron transport chain (ETC) increases life span in many species [1-10]. In C. elegans, mutations in ETC genes such as clk-1 (which encodes a mitochondrial hydroxylase involved in ubiquinone production), isp-1 (which encodes Rieske iron-sulfur protein in complex III), and nuo-6 (which encodes a subunit of NADH dehydrogenase complex) [1-3, 40] have been shown to extend life span. RNAi knock-down of mitochondrial ETC components also increases the life span of C. elegans, and that of Drosophila as well [6, 9]. In mice, mutants that exhibit decreased mitochondrial respiration such as Surf1−/− (mutation in cytochrome c oxidase) [8] and Mclk1+/− (heterozygous knockout of the mouse homolog of clk-1) have long life spans [7]. These findings support the notion that reduced mitochondrial respiration leads to longevity in mammals and suggest the existence of evolutionarily conserved underlying mechanisms.
Because mitochondria are the major source of ROS, one possible mechanism by which a reduction in mitochondrial respiration influences life span may involve changes in the level of ROS. Historically, ROS were believed to be the key causes of aging [41]. The free radical theory of aging proposed that ROS, which are natural byproducts of mitochondrial respiration, cause damage to macromolecules and organelles such as mitochondria. This mitochondrial damage in turn stimulates increased ROS generation, thus evoking a ‘vicious cycle’ that ultimately leads to deterioration of the cell and the organism [41]. However, several studies have shown that many conditions that increase ROS do not decrease life span, in worms and in mice [42, 43, 48]. Moreover, recent studies have highlighted the beneficial role of ROS in longevity [16, 31, 44-48]. For example, antimycin treatment that blocks mitochondria complex III and thus impairs ETC function, triggers excessive ROS production in the mitochondria [49]. Interestingly, antimycin treatment increases the life span of C. elegans [5]. Consistent with this, several studies have shown that long-lived clk-1, isp-1, and nuo-6 mutant worms have increased ROS levels [16, 31, 50]. Likewise, long-lived Mclk1+/− mice exhibit decreased respiration rates and elevated H2O2 levels [7, 51]. These data raise the intriguing possibility that increased ROS may play a causal role in the longevity conferred by the inhibition of mitochondrial respiration.
The Hekimi group recently addressed this issue directly and showed that increased ROS are required for the longevity of isp-1 and nuo-6 mutants [31]. They reported that isp-1 and nuo-6 mutants have increased superoxide levels, whereas clk-1 mutants have elevated overall ROS levels. N-acetylcysteine (NAC), a well-defined antioxidant, significantly decreased the life span of isp-1 and nuo-6 mutants but had little or no effect on the life span of wild-type animals. In case of clk-1 mutants, NAC treatment had marginal effects on life span [31]. In addition, three groups independently showed that wild-type worms treated with the ROS-generating chemicals juglone or paraquat are long lived [16, 31, 45]. These studies are consistent with the previous finding by Schulz et al. that restriction of glucose metabolism increases life span through elevating ROS and that antioxidant treatment suppresses this longevity [46]. Together, these data suggest that increased ROS generation in mitochondrial respiration-defective mutants promotes longevity in C. elegans.
In contrast to the long-lived mitochondrial respiration mutants described above, a mutant allele of mev-1, which encodes a cytochrome b large subunit in complex II [52, 53], cause a short life span [34, 52]. The mev-1 mutants were initially characterized in an EMS mutagenesis screen to identify mutants that are hypersensitive to paraquat treatment [52], and were subsequently shown that these mutants display increased ROS levels [54]. Studies in our laboratory and by Dingely et al. showed that mev-1 mutants have even higher ROS levels than the long-lived clk-1 or isp-1 mutants [16, 55]. Therefore, it is possible that mev-1 mutants are short lived because of excessive ROS, which is similar to the conditions that decrease life span by treatment of high concentrations of ROS-generating chemicals [16, 45].
The role of HIF-1 in longevity induced by impaired respiration and ROS
How do ROS extend the lifespan of respiration mutants? Consistent with experimental results using cultured mammalian cells, recent studies with model organisms show that HIF-1 is stabilized when ROS levels are increased. Mclk1+/− mice, which display reduced mitochondrial respiration, exhibit elevated ROS levels [51] and increased HIF-1α protein levels under normal oxygen conditions [56]. In addition, the increase in HIF-1 levels caused by RNAi knock-down of Mclk1 was abolished by a H2O2-specific antioxidant peptide targeted to mitochondria [56]. In C. elegans, we showed that defects in mitochondrial respiration elevated ROS levels, which lead to increased HIF-1 activity [16]. We found that clk-1 and isp-1 mutations induced several HIF-1 target genes and that the extended life span promoted by clk-1 and isp-1 mutations requires the HIF-1 transcription factor. Moreover, treating the worms with paraquat led to up-regulation of a HIF-1 target gene in a hif-1-dependent manner [16]. Together these data suggest that mutations affecting mitochondrial respiration lengthen life span by increasing ROS levels and HIF-1activity. A recent paper reported that an E. coli TCA cycle-related mutant that cannot provide reducing equivalents used in the ETC, and thus has reduced respiration, showed an extended chronological life span [57]. Interestingly, the longevity of these mutants required ArcA, which is the functional homolog of eukaryotic HIF [57], suggesting that this longevity regulation is functionally conserved between prokaryotes and eukaryotes.
Several issues regarding the role of HIF-1 in the longevity caused by increased ROS remain to be addressed. First, controversy remains regarding how HIF-1 is activated under low oxygen conditions. One unexpected observation regarding hypoxia is that cells exhibit increased ROS levels under hypoxia [58-60]. Although controversial, several recent studies proposed that mitochondrial complex III is the main source of ROS required for HIF-1 stabilization [61-63]. Under normoxic conditions, growth factors required for vasculogenesis and cytokines required for the maintenance of hematopoietic stem cells stabilize HIF-1, and this stabilization requires ROS [64, 65]. It has been suggested that ROS oxidize Fe2+, the coactivator of the HIF prolyl hydroxylase, to Fe3+ through Fenton's reaction in cultured mammalian cells [66]. Follow-up on our study showing that increased ROS lead to the activation of HIF-1 in C. elegans will be crucial to examine this issue in vivo. Second, what are the HIF-1 target genes that mediate the longevity induced by elevated ROS levels? In cultured mammalian cells, increased ROS levels under hypoxia prolong their replicative life span via stabilization of HIF-1, which leads to up-regulation of human telomerase reverse transcriptase (hTERT) and subsequent extension of telomeres [67, 68]. It will be important to determine which HIF-1 target genes are responsible for paraquat-induced longevity in C. elegans. Third, because the hif-1 mutation only partially suppresses the paraquat-induced longevity and because not all hif-1-dependent hypoxia genes were induced by inhibiting respiration [16], additional genes appear to be required in parallel to the conventional HIF-1 signaling pathway. Identification of these genes will increase our understanding of the mechanisms by which increased ROS extend life span.
Mitochondrial unfolded protein response
In contrast to the mitochondrial respiration mutants, we found that longevity caused by RNAi targeting ETC components showed only a partial dependency on hif-1[16]. Together with findings that the effect of respiratory-chain RNAi and respiration mutants may exert different outputs in gene expression and metabolism [11, 13], these studies suggest that there are additional mechanisms by which impaired ETC function (in particular that induced by RNAi) increases life span. Recently, several groups showed that inhibition of mitochondrial respiration triggers the mitochondrial unfolded protein response (UPRmito) [40, 69, 70] and UPRmito has been shown to be required for the longevity caused by impaired mitochondrial respiration [69]. An especially striking finding of Durieux et al. is that RNAi against an ETC component in a single tissue induced longevity of the whole animal, and this RNAi effect conveys a cell non-autonomous signal to neighboring tissues to elicit UPRmito [69]. Specifically, Durieux et al. showed that RNAi knock-down of the ETC component cco-1 (cytochrome c oxidase-1 subunit Vb/COX4) in the intestine or neurons is sufficient to extend the life span of C. elegans. In addition, they showed that cco-1 RNAi in neurons causes UPRmito in the intestine and coined the term “mitokine” for the unidentified signaling molecules that presumably relay the signal induced by reduced mitochondrial respiration in one tissue to other tissues [69]. Based on these results, and together with the suggestion that ROS and HIF-1 mediate the longevity conferred by a reduction in mitochondrial respiration, it is tempting to speculate that ROS such as hydrogen peroxide might travel locally and that HIF-1-target genes propagate a signaling cascade in various tissues to promote longevity.
Conclusion
In this research perspective, we reviewed recent findings about the roles of HIF-1 and ROS in the regulation of aging, focusing on their roles in life span extension induced by impaired mitochondrial respiration in C. elegans (Figure 1). Many interesting questions remain unanswered. Which tissues and functional target genes are important in the regulation of aging by HIF-1? How can both up-regulation and down-regulation of HIF-1 promote longevity? What is the molecular mechanism by which mitochondrial ROS stimulates HIF-1 activity? Future studies using C. elegans will be crucial to address these important issues. Since many aging-regulatory processes are conserved between C. elegans and mammals, these studies may also provide insights into the regulatory mechanisms of aging in mammals, including humans. Moreover, in addition to aging, HIF-1 and mitochondrial impairment have been implicated in various human diseases such as cancer, diabetes, and neurodegenerative diseases. Thus, we believe that these future studies will help us better understand the pathophysiology of these diseases.
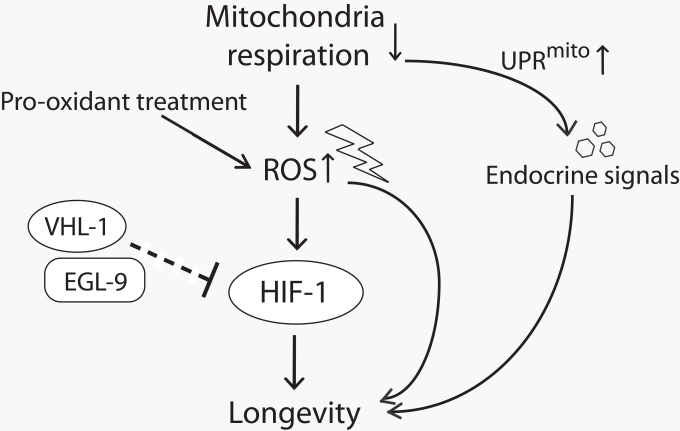
Figure 1. A model for lifespan extension by mild inhibition of mitochondrial respiration in C. elegans Mild reduction in mitochondrial respiration leads to the increase in reactive oxygen species (ROS) production and triggers mitochondrial unfolded protein response (UPRmito). The increased ROS either by reducing mitochondrial respiration or by pro-oxidant treatment promotes longevity at least in part by increasing the activity of hypoxia-inducible factor 1 (HIF-1). EGL-9 (HIF prolyl hydroxylase) and VHL-1 (von Hippel Lindau 1, E3 ubiquitin ligase) affect longevity perhaps through down-regulating HIF-1 (note that this part is currently inconclusive and therefore is shown with a dashed line.). UPRmito caused by impaired mitochondrial respiration extends lifespan through unidentified endocrine signals that relay the longevity response among different tissues.
Acknowledgments
We thank Drs. G-One Ahn and Cynthia Kenyon for helpful comments on the manuscript. This work was supported by WCU (World Class University) program (Project No. R31-2008-000-10100-0) and by the Basic Science Research Program (20100009278) through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology and by the POSTECH Basic Science Research Institute Grant to S.J.L.
References
- 1. Lakowski B and Hekimi S. Determination of life-span in Caenorhabditis elegans by four clock genes. Science. 1996; 272: 1010 -1013. [PubMed] .
- 2. Braeckman BP, Houthoofd K, De Vreese A, Vanfleteren JR. Apparent uncoupling of energy production and consumption in long-lived Clk mutants of Caenorhabditis elegans. Curr Biol. 1999; 9: 493 -496. [PubMed] .
- 3. Feng J, Bussiere F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001; 1: 633 -644. [PubMed] .
- 4. Kirchman PA, Kim S, Lai CY, Jazwinski SM. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics. 1999; 152: 179 -190. [PubMed] .
- 5. Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002; 298: 2398 -2401. [PubMed] .
- 6. Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003; 33: 40 -48. [PubMed] .
- 7. Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005; 19: 2424 -2434. [PubMed] .
- 8. Dell'agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007; 16: 431 -444. [PubMed] .
- 9. Copeland JM, Cho J, Lo T Jr., Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009; 19: 1591 -1598. [PubMed] .
- 10. Kenyon CJ. The genetics of ageing. Nature. 2010; 464: 504 -512. [PubMed] .
- 11. Cristina D, Cary M, Lunceford A, Clarke C, Kenyon C. A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet. 2009; 5: e1000450 [PubMed] .
- 12. Falk MJ, Zhang Z, Rosenjack JR, Nissim I, Daikhin E, Sedensky MM, Yudkoff M, Morgan PG. Metabolic pathway profiling of mitochondrial respiratory chain mutants in C. elegans. Mol Genet Metab. 2008; 93: 388 -397. [PubMed] .
- 13. Butler JA, Ventura N, Johnson TE, Rea SL. Long-lived mitochondrial (Mit) mutants of Caenorhabditis elegans utilize a novel metabolism. FASEB J. 2010; 24: 4977 -4988. [PubMed] .
- 14. Zuryn S, Kuang J, Tuck A, Ebert PR. Mitochondrial dysfunction in Caenorhabditis elegans causes metabolic restructuring, but this is not linked to longevity. Mech Ageing Dev. 2010; 131: 554 -561. [PubMed] .
- 15. Van Raamsdonk JM, Meng Y, Camp D, Yang W, Jia X, Benard C, Hekimi S. Decreased energy metabolism extends life span in Caenorhabditis elegans without reducing oxidative damage. Genetics. 2010; 185: 559 -571. [PubMed] .
- 16. Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010; 20: 2131 -2136. [PubMed] .
- 17. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003; 3: 721 -732. [PubMed] .
- 18. Kaelin WG Jr. and Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008; 30: 393 -402. [PubMed] .
- 19. Leiser SF and Kaeberlein M. The hypoxia-inducible factor HIF-1 functions as both a positive and negative modulator of aging. Biol Chem. 2010; 391: 1131 -1137. [PubMed] .
- 20. Pocock R and Hobert O. Oxygen levels affect axon guidance and neuronal migration in Caenorhabditis elegans. Nat Neurosci. 2008; 11: 894 -900. [PubMed] .
- 21. Packer L and Fuehr K. Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature. 1977; 267: 423 -425. [PubMed] .
- 22. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, Suda T. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010; 7: 391 -402. [PubMed] .
- 23. Fehrer C, Brunauer R, Laschober G, Unterluggauer H, Reitinger S, Kloss F, Gully C, Gassner R, Lepperdinger G. Reduced oxygen tension attenuates differentiation capacity of human mesenchymal stem cells and prolongs their lifespan. Aging Cell. 2007; 6: 745 -757. [PubMed] .
- 24. Kim H, Lee DK, Choi JW, Kim JS, Park SC, Youn HD. Analysis of the effect of aging on the response to hypoxia by cDNA microarray. Mech Ageing Dev. 2003; 124: 941 -949. [PubMed] .
- 25. Frenkel-Denkberg G, Gershon D, Levy AP. The function of hypoxia-inducible factor 1 (HIF-1) is impaired in senescent mice. FEBS Lett. 1999; 462: 341 -344. [PubMed] .
- 26. Leiser SF, Begun A, Kaeberlein M. HIF-1 modulates longevity and healthspan in a temperature-dependent manner. Aging Cell. 2011; .
- 27. Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One. 2009; 4: e6348 [PubMed] .
- 28. Chen D, Thomas EL, Kapahi P. HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 2009; 5: e1000486 [PubMed] .
- 29. Bellier A, Chen CS, Kao CY, Cinar HN, Aroian RV. Hypoxia and the hypoxic response pathway protect against pore-forming toxins in C. elegans. PLoS Pathog. 2009; 5: e1000689 [PubMed] .
- 30. Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science. 2009; 324: 1196 -1198. [PubMed] .
- 31. Yang W and Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010; 8: e1000556 [PubMed] .
- 32. Muller RU, Fabretti F, Zank S, Burst V, Benzing T, Schermer B. The von Hippel Lindau tumor suppressor limits longevity. J Am Soc Nephrol. 2009; 20: 2513 -2517. [PubMed] .
- 33. Jiang H, Guo R, Powell-Coffman JA. The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci U S A. 2001; 98: 7916 -7921. [PubMed] .
- 34. Honda S, Ishii N, Suzuki K, Matsuo M. Oxygen-dependent perturbation of life span and aging rate in the nematode. J Gerontol. 1993; 48: B57 -61. [PubMed] .
- 35. Bishop T, Lau KW, Epstein AC, Kim SK, Jiang M, O'Rourke D, Pugh CW, Gleadle JM, Taylor MS, Hodgkin J, Ratcliffe PJ. Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol. 2004; 2: e289 [PubMed] .
- 36. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001; 107: 43 -54. [PubMed] .
- 37. Klass MR. Aging in the nematode Caenorhabditis elegans: major biological and environmental factors influencing life span. Mech Ageing Dev. 1977; 6: 413 -429. [PubMed] .
- 38. Lee SJ and Kenyon C. Regulation of the longevity response to temperature by thermosensory neurons in Caenorhabditis elegans. Curr Biol. 2009; 19: 715 -722. [PubMed] .
- 39. Treinin M, Shliar J, Jiang H, Powell-Coffman JA, Bromberg Z, Horowitz M. HIF-1 is required for heat acclimation in the nematode Caenorhabditis elegans. Physiol Genomics. 2003; 14: 17 -24. [PubMed] .
- 40. Yang W and Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell. 2010; 9: 433 -447. [PubMed] .
- 41. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956; 11: 298 -300. [PubMed] .
- 42. Perez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y, Richardson A. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009; 1790: 1005 -1014. [PubMed] .
- 43. Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, Matscheski A, Vanfleteren JR, Gems D. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008; 22: 3236 -3241. [PubMed] .
- 44. Ristow M and Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp Gerontol. 2010; 45: 410 -418. [PubMed] .
- 45. Heidler T, Hartwig K, Daniel H, Wenzel U. Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS-generator is dependent on DAF-16 and SIR-2.1. Biogerontology. 2010; 11: 183 -195. [PubMed] .
- 46. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007; 6: 280 -293. [PubMed] .
- 47. Lapointe J and Hekimi S. When a theory of aging ages badly. Cell Mol Life Sci. 2010; 67: 1 -8. [PubMed] .
- 48. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 49. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003; 552: 335 -344. [PubMed] .
- 50. Yang YY, Gangoiti JA, Sedensky MM, Morgan PG. The effect of different ubiquinones on lifespan in Caenorhabditis elegans. Mech Ageing Dev. 2009; 130: 370 -376. [PubMed] .
- 51. Lapointe J and Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J Biol Chem. 2008; 283: 26217 -26227. [PubMed] .
- 52. Ishii N, Takahashi K, Tomita S, Keino T, Honda S, Yoshino K, Suzuki K. A methyl viologen-sensitive mutant of the nematode Caenorhabditis elegans. Mutat Res. 1990; 237: 165 -171. [PubMed] .
- 53. Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998; 394: 694 -697. [PubMed] .
- 54. Senoo-Matsuda N, Yasuda K, Tsuda M, Ohkubo T, Yoshimura S, Nakazawa H, Hartman PS, Ishii N. A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. J Biol Chem. 2001; 276: 41553 -41558. [PubMed] .
- 55. Dingley S, Polyak E, Lightfoot R, Ostrovsky J, Rao M, Greco T, Ischiropoulos H, Falk MJ. Mitochondrial respiratory chain dysfunction variably increases oxidant stress in Caenorhabditis elegans. Mitochondrion. 2010; 10: 125 -136. [PubMed] .
- 56. Wang D, Malo D, Hekimi S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/− mouse mutants. J Immunol. 2010; 184: 582 -590. [PubMed] .
- 57. Gonidakis S, Finkel SE, Longo VD. Genome-wide screen identifies Escherichia coli TCA-cycle-related mutants with extended chronological lifespan dependent on acetate metabolism and the hypoxia-inducible transcription factor ArcA. Aging Cell. 2010; 9: 868 -881. [PubMed] .
- 58. Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998; 95: 11715 -11720. [PubMed] .
- 59. Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998; 273: 18092 -18098. [PubMed] .
- 60. Waypa GB, Marks JD, Mack MM, Boriboun C, Mungai PT, Schumacker PT. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ Res. 2002; 91: 719 -726. [PubMed] .
- 61. Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005; 1: 401 -408. [PubMed] .
- 62. Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005; 1: 393 -399. [PubMed] .
- 63. Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007; 177: 1029 -1036. [PubMed] .
- 64. Patten DA, Lafleur VN, Robitaille GA, Chan DA, Giaccia AJ, Richard DE. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: the essential role of mitochondrial-derived reactive oxygen species. Mol Biol Cell. 2010; 21: 3247 -3257. [PubMed] .
- 65. Yoshida K, Kirito K, Yongzhen H, Ozawa K, Kaushansky K, Komatsu N. Thrombopoietin (TPO) regulates HIF-1alpha levels through generation of mitochondrial reactive oxygen species. Int J Hematol. 2008; 88: 43 -51. [PubMed] .
- 66. Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004; 118: 781 -794. [PubMed] .
- 67. Bell EL, Klimova TA, Eisenbart J, Schumacker PT, Chandel NS. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol Cell Biol. 2007; 27: 5737 -5745. [PubMed] .
- 68. Minamino T, Mitsialis SA, Kourembanas S. Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol Cell Biol. 2001; 21: 3336 -3342. [PubMed] .
- 69. Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011; 144: 79 -91. [PubMed] .
- 70. Ventura N and Rea SL. Caenorhabditis elegans mitochondrial mutants as an investigative tool to study human neurodegenerative diseases associated with mitochondrial dysfunction. Biotechnol J. 2007; 2: 584 -595. [PubMed] .