



In the last week paper in Science Transl Med, Francis Collins, Dimitri Krainc, Kan Cao and co-workers described that rapamycin reverses cellular phenotypes in Hutchinson-Gilford progeria syndrome (HGPS) cells [1]. Is it a co-incidence that rapamycin also suppresses senescence in regular (non-HGPS) mammalian cells [2]?
Clearance of progerin by rapamycin
Hutchinson-Gilford progeria syndrome (HGPS) is a rare genetic disorder characterized by some features reminiscent of aging, including atherosclerosis and alopecia [3-6]. The median life span is 13 years, and the main cause of death is myocardial infarction and stroke. Progeria is mainly caused by the abnormal accumulation of progerin, a mutant form of the nuclear envelope component lamin A [7, 8]. In cell culture, HGPS cells are prone to replicative senescence (Figure 1) [9-12]. Accumulation of progerin causes nuclear abnormalities, mitotic abnormalities and accelerate telomere shortening. This causes DNA damage response, p53 induction and cell cycle arrest [11, 13-16]. After a number of cell divisions in culture, cells stop proliferating (replicative senescence).
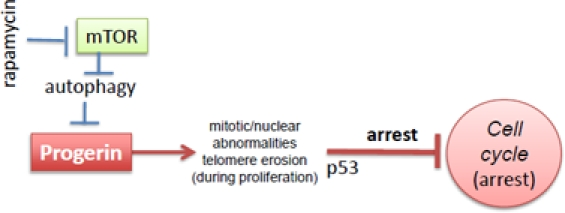
Figure 1. Progerin-induced replicative limit in progeric cells Rapamycin decreases levels of progerin and thus prevents telomere erosion and cell cycle arrest
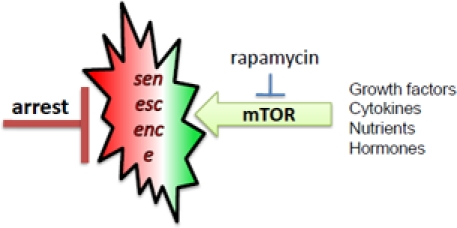
Figure 2. mTOR-driven senescence in arrested normal cells When cell cycle is blocked, mTOR drives senescence
Very recently Cao et al described that rapamycin stimulates clearance of progerin and therefore prevents nuclear abnormalities and delays replicative senescence [1]. Rapamycin eliminates the cause of the abnormalities and therefore is expected to be an effective treatment in progeria [1]. In addition to this strong rationale [1], there is one additional indication for rapamycin, regardless of progerin clearance, namely suppression of geroconversion (conversion from quiescence to senescence) by rapamycin [2, 17-19].
Prevention of geroconversion by rapamycin
Normal human cells undergo replicative senescence due to telomere shortening, which causes cell cycle arrest [9, 20-22]. But cell cycle arrest is not yet senescence [23, 19]. In the young organism, post-mitotic cells are not senescent. Such cells must undergo geroconversion during lifespan. In cell culture, senescence is characterized by large flat cell morphology (hypertrophy), beta-Gal staining, hyper-secretory phenotype, activated DNA-damage response (even in the absence of DNA damage), resistance to signals (such as insulin), elevated cyclins with inappropriate drive into S-phase, and loss of proliferative potential (PP) [17, 24-42, 17]. Loss of PP, a convenient marker in cell culture, means that the cells cannot resume proliferation even when they are released from arrest, for example, by switching p21 off [2, 43]. Senescent phenotype (including loss of PP) can be linked to hyper-active growth-promoting and nutrient-sensing pathways such as mTOR (Target of Rapamycin). In proliferating cells, growth factors (and nutrients) activate cell mass growth, which is balanced by division. When the cell cycle is arrested, then activated mTOR drives the senescent morphology [2, 17]. Over-activation of the mTOR pathway causes hyper-activation and exhaustion of stem cells too [44-46].
In replicative senescence of normal human (not rodent) cells, telomere erosion during 20-70 division cycles causes DDR and cell cycle arrest. Telomere erosion is a very peculiar (and time consuming) method to achieve cell cycle arrest. One can accelerate the process. Cell cycle can be directly arrested by DNA damaging drugs, expression of p53, p21, p16 or of constantly hyper-activated Ras/Rak/Akt [47-50]. When the cell cycle is arrested but mTOR is active then cells senesce [17]. Hyper-active Ras, Raf, Akt not only can arrest cycle but additionally activate the mTOR pathway. Rapamycin and also upstream inhibitors of mTOR suppresses geroconversion in different models of premature and physiological senescence in culture [2, 17, 42, 51-55].
By causing cell cycle arrest, p53 puts such cells on the path of senescence. Simultaneously, p53 can inhibit mTOR [56-60] and can suppress geroconversion [61-66, 58]. This may determine a dual role of p53 in aging [63, 67-70].
mTOR stimulates cellular growth and functions [71-74] and cause signal (insulin) resistance by feedback loops [75-82]. Aging cells are over-activated, hyper-functional (for example, hyper-secretory) and secondary signal resistant [18]. This may result in cell malfunctions and even cellular loss (for example, loss of beta-cells in diabetes type II). Hyper-functions, malfunctions and signal-resistance in turn cause age-related diseases from metabolic syndrome, atherosclerosis and hypertension to neurodegeneration and osteroporosis [83-85]. And not coincidentally, rapamycin is indicated for prevention of all age-related diseases including cancer [83-86]. The sum of all diseases determines the risk of death [85]. And aging is defined as increase of accidence of death with age. So if TOR-dependent cellular aging increases the risk of death, then rapamycin must extend life span. Indeed, inhibition of the TOR pathway extends lifespan in span in diverse organisms from yeast to mice [87-97, 46].
Linking progeria to aging: 4 scenarios
There are at least four models, which are not mutually exclusive
Scenario 1. Progerin is detectable in normal cells from normal elderly humans [98]. In normal human fibroblasts, telomere damage during replicative senescence activates progerin production [10]. In theory, progerin can accumulate. In this scenario, normal aging is caused by progerin or at least in some individuals accumulation of progerin is life-limiting. If so, progeria is a truly accelerated aging or at least accelerated component of aging. Still, there is no evidence so far that progerin normally reaches toxic levels. Also telomere erosion is preferentially a cell culture phenomenon. And elevated progerin production was not seen during cellular senescence that does not entail telomere shortening [10]. On the other hand, patients with dyskeratosis congenita, an inherited bone marrow failure syndrome, have very short telomeres [99]. Also, there may be synergy with additional abnormalities such as abnormalities of nuclear pore complex in aging cells [100].
Scenario 2. Normal aging is caused by overactivation of TOR-centric pathways such as mTOR, MAPK and kinases of the DNA damage response (DDR). Progerin can activate DDR [101]. In turn, DDR may activate the mTOR pathway [102]. Noteworthy, cellular senescence of regular cells is characterized by DDR even in the absence of actual DNA damage (pseudo-DDR) [103]. And pseudo-DDR is inhibited by rapamycin [103]. So there is cross talk between mTOR and DDR [104, 105]. Therefore, by activating DDR pathways, progerin might also promote geroconversion.
Scenario 3. mTOR inhibits autophagy and insufficient autophagy is involved in normal aging [106-109]. Rapamycin also causes clearance of aggregation-prone proteins [110]. In progeria, rapamycin activates clearance of progerin thus slowing down the progeric aging. Thus, rapamycin can affect both progeria and normal aging via activation of autophagy of different proteins and structures.
Scenario 4. Two different mTOR activities are responsible for deceleration of normal and progeric aging. In progeria, this is autophagy. In normal aging, this is suppression of cellular hyper-functions, such as hyperfunctions (such as secretion) and hormone-resistance. Rapamycin would be effective in both conditions but by different reasons. In analogy, rapamycin could be used for certain fungal and viral infection, even though they do not cause normal aging.
Accelerated aging
Still, progeria is not accelerated “normal” aging exactly. What is accelerated “normal” aging or accelerated aging, for brevity. If aging is driven by inappropriate activation of nutrient-, hormone- and mitogen-sensing pathways such as mTOR, then nutrients and insulin can accelerate aging. In fact, obesity is associated with all age-related diseases and dramatically shortens life span. This is the accelerated normal aging. As an example, the maximum “years lost life” (YLL) for white men aged 20 to 30 years with a severe level of obesity (BMI >45) is 13 years, representing a 22% reduction in expected remaining life span [111]. As long ago suggested by the Russian endocrinologist Vladimir Dilman, time flies faster in the obese.
This also could be considered from the point of view of a quasi-programmed aging. Aging is not programmed of course but is an aimless continuation of a program of developmental growth [74, 112-115]. And growth is driven in part by mTOR (activated by growth factors and nutrients).
Rapamycin for progeria and (age-related diseases)
Inhibiting farnesylation of progerin by farnesyl transferase inhibitors (FTI) prevents the nuclear blebbing of progeria and has positive effects in animal models [116-122]. Yet, the FTI lonafarnib is a relatively cytotoxic agent with gastrointestinal and hematological dose-limiting toxicities (in cancer patients) [123]. It was shown that insulin-like growth factor 1 extends longevity in a mouse model of human premature aging [124]. However, there is still a long way to clinical applications.
Therefore, rapamycin, a non-toxic prescription drug, is a very attractive option. Also, in addition to clearance of progerin, rapamycin in theory would suppress geroconversion downstream of progerin. Furthermore, rapamycin prevents atherosclerosis in animal models of accelerated atherosclerosis [125, 126] and can prevent atherosclerotic restenosis in humans [127]. And accelerated atherosclerosis is one of the main symptoms of progeria and ultimately the cause of death.
There is a misconception that rapamycin increases chances of infections and cancer. In reality, rapamycin is an effective cancer preventive agent in both animals [128, 96, 129] and humans [130, 131], in part, because it slows down organismal aging [132]. Rapamycin is not a general immunosuppressant, it induces tolerance to transplanted organs (when used in combination with immunosuppressants). “Figuratively, it transforms immunity from aged-type to infant-type” [83]. Rapamycin can actually improve responses to infections as immunostimulator [133, 46, 134]. Furthermore, to treat progeria (as well as age-related diseases in normal aging), rapamycin will be used in lower doses and intermittently, so a few (if any) side effects could be expected.
As discussed [135], an increase of lipids in blood occurs because rapamycin increases lyposysis (like starvation) and simultaneously decreases lipid entry into the tissues, including the arterial wall. Therefore, rapamycin prevents atherosclerosis in animals and humans (despite increased lipids) [125-127]. Needless to say, rapamycin is a clinically approved, non-toxic drug, which is used for many years in high and chronic doses in transplant patients. But what is about treating children? Rapamycin is successfully used for the treatment of TSC syndrome in children [136]. Now is a turn of progeria [1]. And perhaps now is the time for postponing age-related diseases of normal aging in our life time [137].
Acknowledgments
I thank Kan Cao, Jay Caplan, Zoya Demidenko, Vera Gorbunova, Heinz D. Osiewacz for helpful comments on this manuscript.
References
- 1. Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in hutchinson-gilford progeria syndrome cells. Sci Transl Med. 2011; 3: 89ra58 .
- 2. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009; 8: 1888 -1895. [PubMed] .
- 3. Capell BC, Collins FS, Nabel EG. Mechanisms of cardiovascular disease in accelerated aging syndromes. Circ Res. 2007; 101: 13 -26. [PubMed] .
- 4. Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006; 140: 2603 -2624. [PubMed] .
- 5. Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008; 358: 592 -604. [PubMed] .
- 6. Burtner CR and Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 2010; 11: 567 -578. [PubMed] .
- 7. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003; 423: 293 -298. [PubMed] .
- 8. Ho JC, Zhou T, Lai WH, Huang Y, Chan YC, Li X, Wong NL, Li Y, Au KW, Guo D, Xu J, Siu CW, Pei D, Tse HF, Esteban MA. Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging. 2011; 3: 380 -390. [PubMed] .
- 9. Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, Greider CW, Harley CB. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A. 1992; 89: 10114 -10118. [PubMed] .
- 10. Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest. 2011; .
- 11. Benson EK, Lee SW, Aaronson SA. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J Cell Sci. 2010; 123: 2605 -2612. [PubMed] .
- 12. Huang S, Risques RA, Martin GM, Rabinovitch PS, Oshima J. Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Exp Cell Res. 2008; 314: 82 -91. [PubMed] .
- 13. Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, Kennedy BK. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol Biol Cell. 2008; 19: 5238 -5248. [PubMed] .
- 14. Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM, Zhou Z, Rodriguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, Lopez-Otin C. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005; 437: 564 -568. [PubMed] .
- 15. Musich PR and Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging. 2009; 1: 28 -37. [PubMed] .
- 16. Gonzalez-Suarez I, Redwood AB, Gonzalo S. Loss of A-type lamins and genomic instability. Cell Cycle. 2009; 8: 3860 -3865. [PubMed] .
- 17. Demidenko ZN and Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355 -3361. [PubMed] .
- 18. Blagosklonny MV. Aging-suppressants: cellular senescence (hyperactivation) and its pharmacological deceleration. Cell Cycle. 2009; 8: 1883 -7. [PubMed] .
- 19. Blagosklonny MV. Cell cycle arrest is not senescence. Aging. 2011; 3: 94 -101. [PubMed] .
- 20. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998; 279: 349 -352. [PubMed] .
- 21. Wei S and Sedivy JM. Expression of catalytically active telomerase does not prevent premature senescence caused by overexpression of oncogenic Ha-Ras in normal human fibroblasts. Cancer Res. 1999; 59: 1539 -1543. [PubMed] .
- 22. Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003; 22: 4212 -4222. [PubMed] .
- 23. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4: 358 -362. [PubMed] .
- 24. Dimri GP and Campisi J. Molecular and cell biology of replicative senescence. Cold Spring Harb Symp Quant Biol. 1994; 59: 67 -73. [PubMed] .
- 25. Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007; 371: 21 -31. [PubMed] .
- 26. CoppŽ JP, Patil CK, Rodier F, Sun Y, Mu-oz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008; 6: 2853 -2868. [PubMed] .
- 27. Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F, Lithgow GJ, Campisi J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging. 2009; 1: 402 -411. [PubMed] .
- 28. Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009; 11: 973 -979. [PubMed] .
- 29. CoppŽ JP, Kauser K, Campisi J, BeausŽjour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006; 281: 29568 -29574. [PubMed] .
- 30. Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010; 238 -246. [PubMed] .
- 31. Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K, Roninson IB. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999; 59: 3761 -3767. [PubMed] .
- 32. Satyanarayana A, Greenberg RA, Schaetzlein S, Buer J, Masutomi K, Hahn WC, Zimmermann S, Martens U, Manns MP, Rudolph KL. Mitogen stimulation cooperates with telomere shortening to activate DNA damage responses and senescence signaling. Mol Cell Biol. 2004; 24: 5459 -5474. [PubMed] .
- 33. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008; 133: 1019 -1031. [PubMed] .
- 34. Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006; 10: 459 -472. [PubMed] .
- 35. Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008; 133: 1006 -1018. [PubMed] .
- 36. Dulic V, Drullinger LF, Lees E, Reed SI, Stein GH. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc Natl Acad Sci U S A. 1993; 90: 11034 -11038. [PubMed] .
- 37. Wong H and Riabowol K. Differential CDK-inhibitor gene expression in aging human diploid fibroblasts. Exp Gerontol. 1996; 31: 311 -325. [PubMed] .
- 38. Burton DG, Sheerin AN, Ostler EL, Smith K, Giles PJ, Lowe J, Rhys-Williams W, Kipling DG, Faragher RG. Cyclin D1 overexpression permits the reproducible detection of senescent human vascular smooth muscle cells. Ann N Y Acad Sci. 2007; 1119: 20 -31. [PubMed] .
- 39. Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000; 113: Pt 20 3613 -3622. [PubMed] .
- 40. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006; 5: 187 -195. [PubMed] .
- 41. Lou Z, Wei J, Riethman H, Baur JA, Voglauer R, Shay JW, Wright WE. Telomere length regulates ISG15 expression in human cells. Aging. 2009; 1: 608 -621. [PubMed] .
- 42. Pospelova TV, Demidenk ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009; 8: 4112 -4118. [PubMed] .
- 43. Chang BD, Broude EV, Fang J, Kalinichenko TV, Abdryashitov R, Poole JC, Roninson IB. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000; 19: 2165 -2170. [PubMed] .
- 44. Gan B and DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009; 8: 1003 -1006. [PubMed] .
- 45. Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008; 205: 2397 -2408. [PubMed] .
- 46. Chen C, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009; 2: ra75 [PubMed] .
- 47. Sherr CJ and DePinho RA. Cellular senescence: mitotic clock or culture shock? Cell. 2000; 102: 407 -410. [PubMed] .
- 48. Serrano M and Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol. 2001; 13: 748 -53. [PubMed] .
- 49. Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur J Biochem. 2001; 268: 2784 -2791. [PubMed] .
- 50. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007; 130: 223 -233. [PubMed] .
- 51. Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009; 8: 1896 -1900. [PubMed] .
- 52. Demidenko ZN and Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009; 8: 1901 -1904. [PubMed] .
- 53. Demidenko ZN and Blagosklonny MV. Quantifying pharmacologic suppression of cellular senescence: prevention of cellular hypertrophy versus preservation of proliferative potential. Aging. 2009; 1: 1008 -1016. [PubMed] .
- 54. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 9660-4: 9660 -9664. [PubMed] .
- 55. Leontieva OV and Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging. 2010; 2: 924 -935. [PubMed] .
- 56. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005; 102: 8204 -8209. [PubMed] .
- 57. Feng Z and Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010; 20: 427 -434. [PubMed] .
- 58. Chao SK, Horwitz SB, McDaid HM. Insights into 4E-BP1 and p53 mediated regulation of accelerated cell senescence. Oncotarget. 2010; 2: 89 -98. [PubMed] .
- 59. Budanov AV and Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008; 134: 451 -460. [PubMed] .
- 60. Lee JH, Bodmer R, Bier E, Karin M. Sestrins at the crossroad between stress and aging. Aging. 2010; 2: 369 -374. [PubMed] .
- 61. Leontieva OV, Gudkov AV, Blagosklonny MV. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010; 9: 4323 -4327. [PubMed] .
- 62. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging. 2010; 2: 344 -352. [PubMed] .
- 63. Serrano M. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010; 9: 4256 -4257. [PubMed] .
- 64. Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging. 2010; 2: 535 -537. [PubMed] .
- 65. Santoro R and Blandino G. p53: The pivot between cell cycle arrest and senescence. Cell Cycle. 2010; 9: 4262 -4263. [PubMed] .
- 66. Long JS and Ryan KM. p53 and senescence: a little goes a long way. Cell Cycle. 2010; 9: 4050 -4051. .
- 67. de Keizer PL, Laberge RM, Campisi J. p53: Pro-aging or pro-longevity? Aging. 2010; 2: 377 -379. [PubMed] .
- 68. Vigneron A and Vousden KH. p53, ROS and senescence in the control of aging. Aging. 2010; 2: 471 -474. [PubMed] .
- 69. Poyurovsky MV and Prives C. P53 and aging: A fresh look at an old paradigm. Aging. 2010; 2: 380 -382. [PubMed] .
- 70. Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging. 2010; 2: 748 [PubMed] .
- 71. Schmelzle T and Hall MN. TOR, a central controller of cell growth. Cell. 2000; 103: 253 -262. [PubMed] .
- 72. Edinger AL and Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell. 2002; 13: 2276 -2288. [PubMed] .
- 73. Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005; 17: 596 -603. [PubMed] .
- 74. Blagosklonny MV and Hall MN. Growth and Aging: a common molecular mechanism. Aging. 2009; 1: 357 -362. [PubMed] .
- 75. Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003; 112: 1223 -1233. [PubMed] .
- 76. Tremblay F and Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001; 276: 38052 -38060. [PubMed] .
- 77. Manning BD, Logsdon MN, Lipovsky A, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005; 19: 1773 -1778. [PubMed] .
- 78. Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005; 30: 35 -42. [PubMed] .
- 79. Mordier S and Iynedjian PB. Activation of mammalian target of rapamycin complex 1 and insulin resistance induced by palmitate in hepatocytes. Biochem Biophys Res Commun. 2007; 362: 206 -211. [PubMed] .
- 80. Pani G. P66SHC and ageing: ROS and TOR? Aging. 2010; 2: 514 -518. [PubMed] .
- 81. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011; 332: 1317 -1322. [PubMed] .
- 82. Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, Blenis J. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011; 332: 1322 -1326. [PubMed] .
- 83. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5: 2087 -2102. [PubMed] .
- 84. Blagosklonny MV. Prevention of cancer by inhibiting aging. Cancer Biol Ther. 2008; 7: 1520 -1524. [PubMed] .
- 85. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging. 2009; 1: 281 -288. [PubMed] .
- 86. Tsang CK, Qi H, Liu LF, Zheng XFS. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Disc Today. 2007; 12: 112 -124. .
- 87. Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003; 426: 620 [PubMed] .
- 88. Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004; 14: 885 -890. [PubMed] .
- 89. Powers RWr, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006; 20: 174 -184. [PubMed] .
- 90. Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 2007; 6: 111 -119. [PubMed] .
- 91. Pan Y and Shadel GS. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging. 2009; 1: 131 -145. [PubMed] .
- 92. Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E., Batterham R.L., Kozma S.C., Thomas G., Carling D., Okkenhaug K., Thornton J.M., Partridge L., Gems D., Withers D.J.. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009; 326: 140 -144. [PubMed] .
- 93. Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S, Kapahi P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009; 139: 149 -160. [PubMed] .
- 94. Moskalev AA and Shaposhnikov MV. Pharmacological Inhibition of Phosphoinositide 3 and TOR Kinases Improves Survival of Drosophila melanogaster. Rejuvenation Res. 2009; 13: 246 -7. [PubMed] .
- 95. Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 11: 35 -46. [PubMed] .
- 96. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandezr E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogenous mice. Nature. 2009; 460: 392 -396. [PubMed] .
- 97. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Am J Pathol. 2010; 176: 2092 -2097. [PubMed] .
- 98. McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One. 2007; 2: e1269 [PubMed] .
- 99. Gadalla SM, Cawthon R, Giri N, Alter BP, Savage SA. Telomere length in blood, buccal cells, and fibroblasts from patients with inherited bone marrow failure syndromes. Aging. 2010; 2: 867 -874. [PubMed] .
- 100. Hetzer MW. The role of the nuclear pore complex in aging of post-mitotic cells. Aging. 2010; 2: 74 -75. [PubMed] .
- 101. Manju K, Muralikrishna B, Parnaik VK. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J Cell Sci. 2006; 119: 2704 -2714. [PubMed] .
- 102. Brenneisen P, Wenk J, Wlaschek M, Krieg T, Scharffetter-Kochanek K. Activation of p70 ribosomal protein S6 kinase is an essential step in the DNA damage-dependent signaling pathway responsible for the ultraviolet B-mediated increase in interstitial collagenase (MMP-1) and stromelysin-1 (MMP-3) protein levels in human dermal fibroblasts. J Biol Chem. 2000; 275: 4336 -4344. [PubMed] .
- 103. Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009; 8: 4112 -4118. [PubMed] .
- 104. Rodriguez-Jimenez FJ, Moreno-Manzano V, Mateos-Gregorio P, Royo I, Erceg S, Murguia JR, Sanchez-Puelles JM. FM19G11: A new modulator of HIF that links mTOR activation with the DNA damage checkpoint pathways. Cell Cycle. 2010; 9: 2803 -2813. [PubMed] .
- 105. Alexander A and Walker CL. Differential localization of ATM is correlated with activation of distinct downstream signaling pathways. Cell Cycle. 2010; 9: 3685 -3686. [PubMed] .
- 106. Cavallini G, Donati A, Taddei M, Bergamini E. Evidence for Selective Mitochondrial Autophagy and Failure in Aging. Autophagy. 2007; 3: 1 .
- 107. Hands SL, Proud CG, Wyttenbach A. mTOR's role in ageing: protein synthesis or autophagy? Aging. 2009; 1: 586 -597. [PubMed] .
- 108. Morselli E, Galluzzi L, Kepp O, Criollo A, Maiuri MC, Tavernarakis N, Madeo F, Kroemer G. Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol. Aging. 2009; 1: 961 -970. [PubMed] .
- 109. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004; 36: 585 -595. [PubMed] .
- 110. Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, Rubinsztein DC. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006; 15: 433 -442. [PubMed] .
- 111. Fontaine KR, Redden DT, Wang C, Westfall AO, Allison DB. Years of life lost due to obesity. Jama. 2003; 289: 187 -193. [PubMed] .
- 112. Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010; 9: 3151 -3156. [PubMed] .
- 113. Blagosklonny MV. mTOR-driven aging: speeding car without brakes. Cell Cycle. 2009; 8: 4055 -4059. [PubMed] .
- 114. Blagosklonny MV. Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle. 2010; 9: 683 -688. [PubMed] .
- 115. Blagosklonny MV. Rapamycin and quasi-programmed aging: Four years later. Cell Cycle. 2010; 9: 1859 -1862. [PubMed] .
- 116. Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005; 102: 12879 -12884. [PubMed] .
- 117. Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A. 2005; 102: 10291 -10296. [PubMed] .
- 118. Scaffidi P and Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005; 11: 440 -445. [PubMed] .
- 119. Mallampalli MP, Huyer G, Bendale P, Gelb MH, Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005; 102: 14416 -14421. [PubMed] .
- 120. Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006; 311: 1621 -1623. [PubMed] .
- 121. Glynn MW and Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005; 14: 2959 -2969. [PubMed] .
- 122. Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006; 116: 2115 -2121. [PubMed] .
- 123. Ravoet C, Mineur P, Robin V, Debusscher L, Bosly A, Andre M, El Housni H, Soree A, Bron D, Martiat P. Farnesyl transferase inhibitor (lonafarnib) in patients with myelodysplastic syndrome or secondary acute myeloid leukaemia: a phase II study. Ann Hematol. 2008; 87: 881 -885. [PubMed] .
- 124. Ugalde AP, Marino G, Lopez-Otin C. Rejuvenating somatotropic signaling: a therapeutical opportunity for premature aging? Aging. 2010; 2: 1017 -1022. [PubMed] .
- 125. Mueller MA, Beutner F, Teupser D, Ceglarek U, Thiery J. Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR-/- mice despite severe hypercholesterolemia. Atherosclerosis. 2008; 198: 39 -48. [PubMed] .
- 126. Zhao L, Ding T, Cyrus T, Cheng Y, Tian H, Ma M, Falotico R, Pratico D. Low-dose oral sirolimus reduces atherogenesis, vascular inflammation and modulates plaque composition in mice lacking the LDL receptor. Br J Pharmacol. 2009; 156: 774 -785. [PubMed] .
- 127. Rodriguez AE, Granada JF, Rodriguez-Alemparte M, Vigo CF, Delgado J, Fernandez-Pereira C, Pocovi A, Rodriguez-Granillo AM, Schulz D, Raizner AE, Palacios I, O'neill W, Kaluza GL, Stone G, Investigators OI. Oral Rapamycin After Coronary Bare-Metal Stent Implantation to Prevent Restenosis The Prospective, Randomized Oral Rapamycin in Argentina (ORAR II) Study. J Am Coll Cardiol. 2006; 47: 1522 -1529. [PubMed] .
- 128. Granville CA, Warfel N, Tsurutani J, Hollander MC, Robertson M, Fox SD, Veenstra TD, Issaq HJ, Linnoila RI, Dennis PA. Identification of a highly effective rapamycin schedule that markedly reduces the size, multiplicity, and phenotypic progression of tobacco carcinogen-induced murine lung tumors. Clin Cancer Res. 2007; 13: 2281 -2289. [PubMed] .
- 129. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176: 2092 -2097. [PubMed] .
- 130. Mathew T, Kreis H, Friend P. Two-year incidence of malignancy in sirolimus-treated renal transplant recipients: results from five multicenter studies. Clin Transplant. 2004; 18: 446 -449. [PubMed] .
- 131. Kauffman HM, Cherikh WS, Cheng Y, Hanto DW, Kahan BD. Maintenance immunosuppression with target-of-rapamycin inhibitors is associated with a reduced incidence of de novo malignancies. Transplantation. 2005; 80: 883 -889. [PubMed] .
- 132. Blagosklonny MV. Prevention of cancer by inhibiting aging. Cancer Biol Ther. 2008; 7: 1520 -1524. [PubMed] .
- 133. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009; 460: 108 -112. [PubMed] .
- 134. Rao RR, Li Q, Shrikant PA. Fine-tuning CD8(+) T cell functional responses: mTOR acts as a rheostat for regulating CD8(+) T cell proliferation, survival and differentiation? Cell Cycle. 2010; 9: 2996 -3001. [PubMed] .
- 135. Blagosklonny MV. An anti-aging drug today: from senescence-promoting genes to anti-aging pill. Drug Disc Today. 2007; 12: 218 -224. .
- 136. Major P. Potential of mTOR inhibitors for the treatment of subependymal giant cell astrocytomas in tuberous sclerosis complex. Aging. 2011; 3: 189 -191. [PubMed] .
- 137. Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: Preliminary proposal. Cell Cycle. 2010; 2: 4788 -4794. [PubMed] .